
【數(shù)據(jù)競賽】天池-蛋白質(zhì)結(jié)構(gòu)預(yù)測大賽冠軍方案分享
1. 賽題介紹
賽題鏈接:https://tianchi.aliyun.com/competition/entrance/231781/introduction
本題為根據(jù)蛋白質(zhì)的一級結(jié)構(gòu)預(yù)測其二級結(jié)構(gòu),經(jīng)過比賽期間組內(nèi)師兄的講解,我對蛋白質(zhì)一級結(jié)構(gòu)二級結(jié)構(gòu)的理解如下,如有錯(cuò)誤,歡迎指正。
蛋白質(zhì)可以看成是一條氨基酸序列,在空間中是一種相互交錯(cuò)螺旋的結(jié)構(gòu),像一條互相纏繞的繩子:
這種三維結(jié)構(gòu)叫做蛋白質(zhì)的三級結(jié)構(gòu),而如果不考慮結(jié)構(gòu)的三維性,或者說把這整條序列拉直,用一個(gè)一維的序列表示,這便是得到了蛋白質(zhì)的一級結(jié)構(gòu):
GPTGTGESKCPLMVKVLDAV······
這些字母G、A、V等便是代表一個(gè)個(gè)的氨基酸,其中主要包含有20種常見的氨基酸。
用這樣的序列表示蛋白質(zhì)比起原始的三維結(jié)構(gòu)確實(shí)方便不少,但卻丟失了三維的結(jié)構(gòu)信息,蛋白質(zhì)的結(jié)構(gòu)決定其功能,這里的結(jié)構(gòu)不止是序列本身,更多的還依賴其三維結(jié)構(gòu)。因此,便出現(xiàn)了蛋白質(zhì)的二級結(jié)構(gòu),它是一條與一級結(jié)構(gòu)長度相等的一維序列,用以表征一級結(jié)構(gòu)種的各位置的氨基酸在三維空間種的形態(tài),以保留一部分的三維結(jié)構(gòu)信息,例如以上蛋白質(zhì)一節(jié)結(jié)構(gòu)對應(yīng)的二級結(jié)構(gòu)為:
EEEEEEETT······
==注意:以上二級結(jié)構(gòu)最前面有11個(gè)空白符,這個(gè)空白符也是在蛋白質(zhì)在三維空間種的一種松散結(jié)構(gòu)表示。==
這里的' '、'E'、'T'等都是對應(yīng)位置的氨基酸在空間種的形態(tài)(與一級結(jié)構(gòu) GPTGTGESKCPLMVKVLDAV······ 是一一對應(yīng)的),例如'T'代表的就是該位置的氨基酸在空間中是一種氫鍵轉(zhuǎn)折的形態(tài)。
本賽題就是需要通過蛋白質(zhì)的一級結(jié)構(gòu),預(yù)測其二級結(jié)構(gòu),在深度學(xué)習(xí)種是一種典型的N-N的seq2seq問題。
2. 賽題理解
不難想到,蛋白質(zhì)三維結(jié)構(gòu)的形成,其實(shí)主要是受某些力的作用,不同氨基酸的分子量、體積、質(zhì)量等性質(zhì)都有差異,這些小分子間會受到分子間作用力的影響,換句話說,分子間作用力等多種因素共同作用,讓蛋白質(zhì)形成了這樣的一種相對穩(wěn)定的空間結(jié)構(gòu),以達(dá)到一種穩(wěn)態(tài);而倘若你強(qiáng)行把它拉直,它也會由于受力不均,又開始相互纏繞,以達(dá)到穩(wěn)態(tài)。
因此,對于某條蛋白質(zhì)的二級結(jié)構(gòu)中第i個(gè)位置的空間形態(tài),其不止是取決于對應(yīng)一級結(jié)構(gòu)中位置i的氨基酸,還取決于位置i周圍氨基酸甚至整條序列的情況。
定義一級結(jié)構(gòu)中位置i及其上下文的整條片段為X,對應(yīng)的二級結(jié)構(gòu)中位置i的形態(tài)為Y,我統(tǒng)計(jì)了整個(gè)訓(xùn)練數(shù)據(jù)中 P(Y|X) 的情況,并計(jì)算了在不同窗口大小時(shí),P(Y|X)>0.95 在所有 P(Y|X) 中的占比情況如下表:
| 窗口大小 | 1 | 3 | 5 | 7 | 9 | 13 | … |
|---|---|---|---|---|---|---|---|
| P(P(Y|X)>0.95) | 0.00588 | 0.02188 | 0.55728 | 0.83511 | 0.84413 | 0.85431 | … |
以上結(jié)果也驗(yàn)證了之前的理解,且不難看出,當(dāng)窗口大小達(dá)到7以上時(shí),可以達(dá)到較好的預(yù)測。
3. 思路分享
這類題首先需要解決的是輸入序列的編碼問題,很自然的可以想到onehot和word2vec兩種編碼方法,本次賽題我們都進(jìn)行了嘗試。
3.1 Onehot與基本理化性質(zhì)編碼+滑窗法+淺層NN
氨基酸的基本理化性質(zhì)包括分子量、等電點(diǎn)、解離常數(shù)、范德華半徑、水中溶解度、側(cè)臉疏水性,以及形成α螺旋可能性、形成β螺旋可能性、轉(zhuǎn)向概率等(來自Chou-Fasman算法),這些數(shù)據(jù)百度都很容易找到。
然后是窗口大小的選擇。經(jīng)過測試,隱層節(jié)點(diǎn)數(shù)為1024,當(dāng)窗口大小達(dá)到79以上時(shí),線下MaF達(dá)到飽和,為0.749。再調(diào)節(jié)隱層大小為2048,最后的線下MaF為0.767。
==(注:此處為氨基酸級別的MaF得分,非官方評分方法的序列級MaF再平均的結(jié)果;且此處未進(jìn)行交叉驗(yàn)證,僅僅是單模的結(jié)果,后面的線上結(jié)果也是,所以可能會有一定偏差)。==
該模型提交后線上結(jié)果為0.7312。(滑窗模型其實(shí)等價(jià)于基于整條序列的CNN模型)
3.2 Word2vec+深層NN
NN的結(jié)構(gòu)設(shè)計(jì)主要參考論文《Protein Secondary Structure Prediction Using Cascaded Convolutional and Recurrent Neural Networks》[1],這是一篇使用深度學(xué)習(xí)進(jìn)行蛋白質(zhì)二級結(jié)構(gòu)預(yù)測的經(jīng)典論文,文中使用了CNN+BiGRU的結(jié)構(gòu)進(jìn)行蛋白質(zhì)二級結(jié)構(gòu)預(yù)測,模型結(jié)構(gòu)如下:
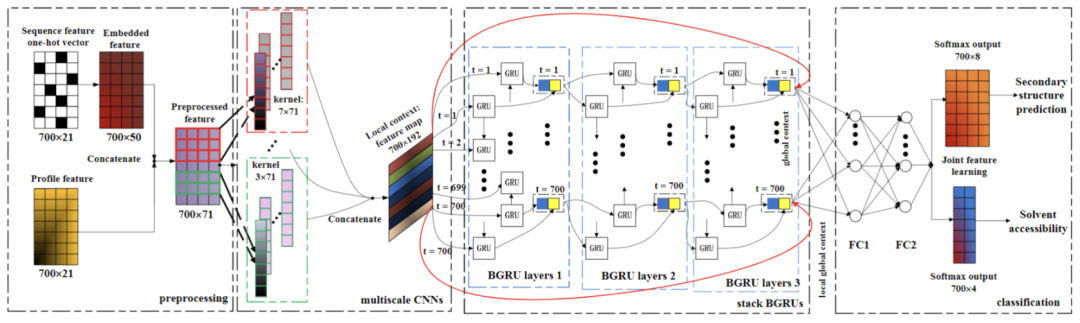
該模型先通過CNN捕獲局部信息,再通過RNN融入全局信息,是NLP長文本任務(wù)的常見baseline模型。這里基本照搬了模型結(jié)構(gòu),但將編碼部分改為了word2vec預(yù)訓(xùn)練的結(jié)構(gòu),詞向量大小為128,其它結(jié)構(gòu)和參數(shù)與原文一致,文章可從github項(xiàng)目目錄進(jìn)行下載。
此處還需注意一定的是,如果embedding層是單獨(dú)對每個(gè)氨基酸進(jìn)行編碼的話,那么詞表大小為23(數(shù)據(jù)集中共23種字母)。而在NLP種經(jīng)常用到的一種叫做n-gram的技術(shù),即將多個(gè)詞綁定在以此形成整體,這個(gè)技術(shù)在蛋白質(zhì)序列種也用得比較多,成為k-mers。倘若使用k-mers構(gòu)建詞表的話,假定k=3,那么詞表的大小就是232323=12167,這樣相當(dāng)于在編碼時(shí)將上下文也考慮了進(jìn)去,增加了詞的多樣性,在一定程度上可以提高模型的學(xué)習(xí)能力,但也會增大過擬合的風(fēng)險(xiǎn)。
這里我也分別嘗試了k=1和k=3的兩個(gè)模型情況,線下分別為0.719和0.706,線上分別為0.7576和0.7518。
==(注:此處計(jì)算線下分?jǐn)?shù)的方法與官方提供的是一致的,但不知道為何線下比線上低了這么多,暫時(shí)還未找到原因;且此處未進(jìn)行交叉驗(yàn)證,僅僅是單模的結(jié)果,后面的線上結(jié)果也是,所以可能會有一定偏差)==
這兩個(gè)模型的輸入和數(shù)據(jù)劃分都有較大差異,顯然會有一定的融合收益,將二者的結(jié)果進(jìn)行加權(quán)平均后,線上結(jié)果為0.7702。
3.3 最終模型
將以上幾個(gè)模型的特征輸入都有著較大不同,進(jìn)行簡單加權(quán)融合后,線上結(jié)果到達(dá)0.7770。
在得到以上結(jié)果后,進(jìn)一步分析問題:
1.模型真正學(xué)會的到底是什么: 我們結(jié)合賽題理解部分的統(tǒng)計(jì)結(jié)果,不難想到,與其說模型是學(xué)會了推理,不如說模型主要是記憶了大量由X->Y的固定映射或搭配,然后根據(jù)不同搭配的置信度進(jìn)行決策,學(xué)會如何權(quán)衡不同搭配以得到更加正確的結(jié)果,這也是選用單層小窗口CNN時(shí)嚴(yán)重欠擬合的原因。
2.氨基酸的編碼表示: 在NLP任務(wù)中的字詞編碼由于詞表過大導(dǎo)致維度過多且稀疏,這才出現(xiàn)了word2vec算法以得到詞語的低維稠密表示,且不同字詞間有很大的聯(lián)系,而這一聯(lián)系可以通過詞向量間的cos距離等來刻畫。而蛋白質(zhì)總共包括的氨基酸種類較少,在本數(shù)據(jù)中只有23種,只需要一個(gè)23維的onehot向量就可以表示,且不同氨基酸間的關(guān)聯(lián)度很小,更多的是差異性,而onehot向量是可以充分表達(dá)這一差異性的(不同onehot向量在高維空間中相互垂直)。這也是簡單的onehot編碼+大窗口CNN能如此有效的原因,也驗(yàn)證了前面的觀點(diǎn),即模型主要是記憶了大量由X->Y的隱射,預(yù)測時(shí)根據(jù)所記憶的大量先驗(yàn)知識,對輸出進(jìn)行決策。
綜上,我們設(shè)計(jì)了最終的模型:在3.2部分的模型中,將embedding部分改成了25維onehot編碼+14維理化特征+25維word2vec特征,其中onehot和理化特征部分在訓(xùn)練過程中是frozen的,而word2vec會隨著訓(xùn)練進(jìn)行finetune;其次是加大了CNN部分的窗口,設(shè)置成了[1,9,81]。
PS:這部分沒有嘗試其它的數(shù)值,這里是玄學(xué)設(shè)計(jì),取了一個(gè)單粒度的窗口1(相當(dāng)于最普通的神經(jīng)網(wǎng)絡(luò),僅僅是對特征進(jìn)行了非線性變換);大窗口81(為了達(dá)到之前的最優(yōu)窗口79);以及大窗口開根號的數(shù)值——9,以折個(gè)中)
最終按次方案訓(xùn)練了一個(gè)3折的模型,線下MaF平均為0.756。
==(注:此處計(jì)算線下分?jǐn)?shù)的方法是padding后序列級別的MaF再平均,理論上應(yīng)該會高于去掉padding后的結(jié)果,這里同樣線上的結(jié)果也好于線下==
將3折的模型加權(quán)平均后線上分?jǐn)?shù)為0.7832。(榜上最優(yōu)結(jié)果0.7855是融合了之前的幾個(gè)模型,但沒多大參考價(jià)值,最終模型可以說是融合了之前的所有模型,融合沒多大價(jià)值,收益基本來自數(shù)據(jù)的分布差異)
4. 代碼開源
代碼在github進(jìn)行了開源,基于pytorch,其中主要包含:
nnLayer:基本神經(jīng)網(wǎng)絡(luò)結(jié)構(gòu)的封裝。DL_ClassifierModel:整個(gè)模型的封裝,包含訓(xùn)練、模型的加載保存等部分。utils:數(shù)據(jù)接口部分的封裝。metrics:評價(jià)指標(biāo)函數(shù)的封裝。SecondStructurePredictor:模型的預(yù)測接口類。
使用方法如下:
#?導(dǎo)入相關(guān)類
from?utils?import?*
from?DL_ClassifierModel?import?*
from?SecondStructurePredictor?import?*
#?初始化數(shù)據(jù)類
dataClass?=?DataClass('data_seq_train.txt',?'data_sec_train.txt',?k=1,?validSize=0.3,?minCount=0)
#?詞向量預(yù)訓(xùn)練
dataClass.vectorize(method='char2vec',?feaSize=25,?sg=1)
#?onehot+理化特征獲取
dataClass.vectorize(method='feaEmbedding')
#?初始化模型對象
model?=?FinalModel(classNum=dataClass.classNum,?embedding=dataClass.vector['embedding'],?feaEmbedding=dataClass.vector['feaEmbedding'],?
???????????????????useFocalLoss=True,?device=torch.device('cuda'))
#?開始訓(xùn)練
model.cv_train(?dataClass,?trainSize=64,?batchSize=64,?epoch=1000,?stopRounds=100,?earlyStop=30,?saveRounds=1,
????????????????savePath='model/FinalModel',?lr=3e-4,?augmentation=0.1,?kFold=3)
#?預(yù)測, 得到的輸出是一個(gè)N × L × C的矩陣,N為樣例數(shù),L為序列最大長度,C為類別數(shù),即得到的是各序列各位置得到各類別上的概率。
model?=?Predictor_final('model/FinalModelxxx.pkl',?device='xxx',?map_location='xxx')
model.predict('seqData.txt',?batchSize=128)
參考文獻(xiàn)
[1]Li Z, Yu Y. Protein secondary structure prediction using cascaded convolutional and recurrent neural networks[J]. arXiv preprint arXiv:1604.07176, 2016.
往期精彩回顧
獲取本站知識星球優(yōu)惠券,復(fù)制鏈接直接打開:
https://t.zsxq.com/qFiUFMV
本站qq群704220115。
加入微信群請掃碼: