WGCNA分析,簡單全面的最新教程
NGS系列文章包括NGS基礎(chǔ)、轉(zhuǎn)錄組分析?(Nature重磅綜述|關(guān)于RNA-seq你想知道的全在這)、ChIP-seq分析?(ChIP-seq基本分析流程)、單細(xì)胞測序分析?(重磅綜述:三萬字長文讀懂單細(xì)胞RNA測序分析的最佳實(shí)踐教程 (原理、代碼和評述))、DNA甲基化分析、重測序分析、GEO數(shù)據(jù)挖掘(典型醫(yī)學(xué)設(shè)計(jì)實(shí)驗(yàn)GEO數(shù)據(jù)分析 (step-by-step) - Limma差異分析、火山圖、功能富集)等內(nèi)容。
WGCNA基本概念
加權(quán)基因共表達(dá)網(wǎng)絡(luò)分析 (WGCNA, Weighted correlation network analysis)是用來描述不同樣品之間基因關(guān)聯(lián)模式的系統(tǒng)生物學(xué)方法,可以用來鑒定高度協(xié)同變化的基因集, 并根據(jù)基因集的內(nèi)連性和基因集與表型之間的關(guān)聯(lián)鑒定候補(bǔ)生物標(biāo)記基因或治療靶點(diǎn)。
相比于只關(guān)注差異表達(dá)的基因,WGCNA利用數(shù)千或近萬個(gè)變化最大的基因或全部基因的信息識別感興趣的基因集,并與表型進(jìn)行顯著性關(guān)聯(lián)分析。一是充分利用了信息,二是把數(shù)千個(gè)基因與表型的關(guān)聯(lián)轉(zhuǎn)換為數(shù)個(gè)基因集與表型的關(guān)聯(lián),免去了多重假設(shè)檢驗(yàn)校正的問題。
理解WGCNA,需要先理解下面幾個(gè)術(shù)語和它們在WGCNA中的定義。
共表達(dá)網(wǎng)絡(luò):定義為加權(quán)基因網(wǎng)絡(luò)。點(diǎn)代表基因,邊代表基因表達(dá)相關(guān)性。加權(quán)是指對相關(guān)性值進(jìn)行冥次運(yùn)算(冥次的值也就是軟閾值?(power, pickSoftThreshold這個(gè)函數(shù)所做的就是確定合適的power))。無向網(wǎng)絡(luò)的邊屬性計(jì)算方式為
abs(cor(genex, geney)) ^ power;有向網(wǎng)絡(luò)的邊屬性計(jì)算方式為(1+cor(genex, geney)/2) ^ power; sign hybrid的邊屬性計(jì)算方式為cor(genex, geney)^power if cor>0 else 0。這種處理方式強(qiáng)化了強(qiáng)相關(guān),弱化了弱相關(guān)或負(fù)相關(guān),使得相關(guān)性數(shù)值更符合無標(biāo)度網(wǎng)絡(luò)特征,更具有生物意義。如果沒有合適的power,一般是由于部分樣品與其它樣品因?yàn)槟撤N原因差別太大導(dǎo)致的,可根據(jù)具體問題移除部分樣品或查看后面的經(jīng)驗(yàn)值。Module(模塊):高度內(nèi)連的基因集。在無向網(wǎng)絡(luò)中,模塊內(nèi)是高度相關(guān)的基因。在有向網(wǎng)絡(luò)中,模塊內(nèi)是高度正相關(guān)的基因。把基因聚類成模塊后,可以對每個(gè)模塊進(jìn)行三個(gè)層次的分析:
1. 功能富集分析查看其功能特征是否與研究目的相符;2. 模塊與性狀進(jìn)行關(guān)聯(lián)分析,找出與關(guān)注性狀相關(guān)度最高的模塊;3. 模塊與樣本進(jìn)行關(guān)聯(lián)分析,找到樣品特異高表達(dá)的模塊。基因富集相關(guān)文章?去東方,最好用的在線GO富集分析工具;GO、GSEA富集分析一網(wǎng)打進(jìn);GSEA富集分析-界面操作。其它關(guān)聯(lián)后面都會(huì)提及。
Connectivity (連接度):類似于網(wǎng)絡(luò)中 “度” (degree)的概念。每個(gè)基因的連接度是與其相連的基因的
邊屬性之和。Module eigengene E: 給定模型的第一主成分,代表整個(gè)模型的基因表達(dá)譜。這個(gè)是個(gè)很巧妙的梳理,我們之前講過PCA分析的降維作用,之前主要是拿來做可視化,現(xiàn)在用到這個(gè)地方,很好的用一個(gè)向量代替了一個(gè)矩陣,方便后期計(jì)算。(降維除了PCA,還可以看看tSNE)
Intramodular connectivity: 給定基因與給定模型內(nèi)其他基因的關(guān)聯(lián)度,判斷基因所屬關(guān)系。
Module membership: 給定基因表達(dá)譜與給定模型的eigengene的相關(guān)性。
Hub gene: 關(guān)鍵基因 (連接度最多或連接多個(gè)模塊的基因)。
Adjacency matrix (鄰接矩陣):基因和基因之間的加權(quán)相關(guān)性值構(gòu)成的矩陣。
TOM (Topological overlap matrix):把鄰接矩陣轉(zhuǎn)換為拓?fù)渲丿B矩陣,以降低噪音和假相關(guān),獲得的新距離矩陣,這個(gè)信息可拿來構(gòu)建網(wǎng)絡(luò)或繪制TOM圖。
基本分析流程
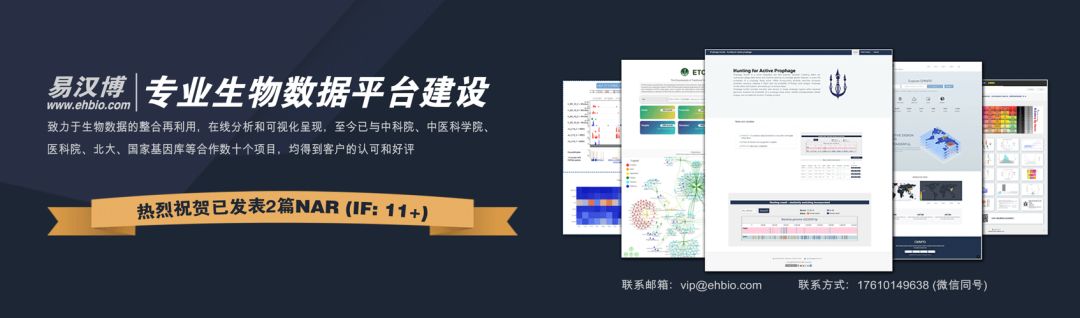
構(gòu)建基因共表達(dá)網(wǎng)絡(luò):使用加權(quán)的表達(dá)相關(guān)性。
識別基因集:基于加權(quán)相關(guān)性,進(jìn)行層級聚類分析,并根據(jù)設(shè)定標(biāo)準(zhǔn)切分聚類結(jié)果,獲得不同的基因模塊,用聚類樹的分枝和不同顏色表示。
如果有表型信息,計(jì)算基因模塊與表型的相關(guān)性,鑒定性狀相關(guān)的模塊。
研究模型之間的關(guān)系,從系統(tǒng)層面查看不同模型的互作網(wǎng)絡(luò)。
從關(guān)鍵模型中選擇感興趣的驅(qū)動(dòng)基因,或根據(jù)模型中已知基因的功能推測未知基因的功能。
導(dǎo)出TOM矩陣,繪制相關(guān)性圖。
WGCNA包實(shí)戰(zhàn)
R包WGCNA是用于計(jì)算各種加權(quán)關(guān)聯(lián)分析的功能集合,可用于網(wǎng)絡(luò)構(gòu)建,基因篩選,基因簇鑒定,拓?fù)涮卣饔?jì)算,數(shù)據(jù)模擬和可視化等。
輸入數(shù)據(jù)和參數(shù)選擇
WGCNA本質(zhì)是基于相關(guān)系數(shù)的網(wǎng)絡(luò)分析方法,適用于多樣品數(shù)據(jù)模式,一般要求樣本數(shù)多于15個(gè)。樣本數(shù)多于20時(shí)效果更好,樣本越多,結(jié)果越穩(wěn)定。
基因表達(dá)矩陣: 常規(guī)表達(dá)矩陣即可,即基因在行,樣品在列,進(jìn)入分析前做一個(gè)轉(zhuǎn)置。RPKM、FPKM或其它標(biāo)準(zhǔn)化方法影響不大,推薦使用Deseq2的
varianceStabilizingTransformation或log2(x+1)對標(biāo)準(zhǔn)化后的數(shù)據(jù)做個(gè)轉(zhuǎn)換。如果數(shù)據(jù)來自不同的批次,需要先移除批次效應(yīng) (記得上次轉(zhuǎn)錄組培訓(xùn)課講過如何操作)。如果數(shù)據(jù)存在系統(tǒng)偏移,需要做下quantile normalization。性狀矩陣:用于關(guān)聯(lián)分析的性狀必須是數(shù)值型特征 (如下面示例中的
Height,?Weight,Diameter)。如果是區(qū)域或分類變量,需要轉(zhuǎn)換為0-1矩陣的形式(1表示屬于此組或有此屬性,0表示不屬于此組或無此屬性,如樣品分組信息WT, KO, OE)。ID ?WT ?KO ?OE Height Weight Diameter
samp1 ? 1 ? 0 ? 0 ? 1 ? 2 ? 3
samp2 ? 1 ? 0 ? 0 ? 2 ? 4 ? 6
samp3 ? 0 ? 1 ? 0 ? 10 ?20 ?50
samp4 ? 0 ? 1 ? 0 ? 15 ?30 ?80
samp5 ? 0 ? 0 ? 1 ? NA ?9 ? 8
samp6 ? 0 ? 0 ? 1 ? 4 ? 8 ? 7推薦使用
Signed network和Robust correlation (bicor)。(這個(gè)根據(jù)自己的需要,看看上面寫的每個(gè)網(wǎng)絡(luò)怎么計(jì)算的,更知道怎么選擇)無向網(wǎng)絡(luò)在power小于
15或有向網(wǎng)絡(luò)power小于30內(nèi),沒有一個(gè)power值可以使無標(biāo)度網(wǎng)絡(luò)圖譜結(jié)構(gòu)R^2達(dá)到0.8或平均連接度降到100以下,可能是由于部分樣品與其他樣品差別太大造成的。這可能由批次效應(yīng)、樣品異質(zhì)性或實(shí)驗(yàn)條件對表達(dá)影響太大等造成, 可以通過繪制樣品聚類查看分組信息、關(guān)聯(lián)批次信息、處理信息和有無異常樣品 (可以使用之前講過的熱圖簡化,增加行或列屬性)。如果這確實(shí)是由有意義的生物變化引起的,也可以使用后面程序中的經(jīng)驗(yàn)power值。
安裝WGCNA
WGCNA依賴的包比較多,bioconductor上的包需要自己安裝,cran上依賴的包可以自動(dòng)安裝。通常在R中運(yùn)行下面4條語句就可以完成WGCNA的安裝。
建議在編譯安裝R時(shí)增加--with-blas --with-lapack提高矩陣運(yùn)算的速度,具體見R和Rstudio安裝。
#source("https://bioconductor.org/biocLite.R")
#biocLite(c("AnnotationDbi", "impute","GO.db", "preprocessCore"))
#site="https://mirrors.tuna.tsinghua.edu.cn/CRAN"
#install.packages(c("WGCNA", "stringr", "reshape2"), repos=site)WGCNA實(shí)戰(zhàn)
實(shí)戰(zhàn)采用的是官方提供的清理后的矩陣,原矩陣信息太多,容易給人誤導(dǎo),后臺回復(fù)WGCNA?獲取數(shù)據(jù)。
數(shù)據(jù)讀入
library(WGCNA)
## Loading required package: dynamicTreeCut
## Loading required package: fastcluster
##
## Attaching package: 'fastcluster'
## The following object is masked from 'package:stats':
##
## ? ? hclust
## ==========================================================================
## *
## * ?Package WGCNA 1.63 loaded.
## *
## * ? ?Important note: It appears that your system supports multi-threading,
## * ? ?but it is not enabled within WGCNA in R.
## * ? ?To allow multi-threading within WGCNA with all available cores, use
## *
## * ? ? ? ? ?allowWGCNAThreads()
## *
## * ? ?within R. Use disableWGCNAThreads() to disable threading if necessary.
## * ? ?Alternatively, set the following environment variable on your system:
## *
## * ? ? ? ? ?ALLOW_WGCNA_THREADS=
## *
## * ? ?for example
## *
## * ? ? ? ? ?ALLOW_WGCNA_THREADS=48
## *
## * ? ?To set the environment variable in linux bash shell, type
## *
## * ? ? ? ? ? export ALLOW_WGCNA_THREADS=48
## *
## * ? ? before running R. Other operating systems or shells will
## * ? ? have a similar command to achieve the same aim.
## *
## ==========================================================================
##
## Attaching package: 'WGCNA'
## The following object is masked from 'package:stats':
##
## ? ? cor
library(reshape2)
library(stringr)
#
options(stringsAsFactors = FALSE)
# 打開多線程
enableWGCNAThreads()
## Allowing parallel execution with up to 47 working processes.
# 常規(guī)表達(dá)矩陣,log2轉(zhuǎn)換后或
# Deseq2的varianceStabilizingTransformation轉(zhuǎn)換的數(shù)據(jù)
# 如果有批次效應(yīng),需要事先移除,可使用removeBatchEffect
# 如果有系統(tǒng)偏移(可用boxplot查看基因表達(dá)分布是否一致),
# 需要quantile normalization
exprMat <- "WGCNA/LiverFemaleClean.txt"
# 官方推薦 "signed" 或 "signed hybrid"
# 為與原文檔一致,故未修改
type = "unsigned"
# 相關(guān)性計(jì)算
# 官方推薦 biweight mid-correlation & bicor
# corType: pearson or bicor
# 為與原文檔一致,故未修改
corType = "pearson"
corFnc = ifelse(corType=="pearson", cor, bicor)
# 對二元變量,如樣本性狀信息計(jì)算相關(guān)性時(shí),
# 或基因表達(dá)嚴(yán)重依賴于疾病狀態(tài)時(shí),需設(shè)置下面參數(shù)
maxPOutliers = ifelse(corType=="pearson",1,0.05)
# 關(guān)聯(lián)樣品性狀的二元變量時(shí),設(shè)置
robustY = ifelse(corType=="pearson",T,F)
##導(dǎo)入數(shù)據(jù)##
dataExpr <- read.table(exprMat, sep='\t', row.names=1, header=T,
? ? ? ? ? ? ? ? ? ? quote="", comment="", check.names=F)
dim(dataExpr)
## [1] 3600 ?134
head(dataExpr)[,1:8]
## ? ? ? ? ? ? ? ? F2_2 ? ?F2_3 ? ? F2_14 ? ?F2_15 ? ?F2_19 ? ? ? F2_20
## MMT00000044 -0.01810 ?0.0642 ?6.44e-05 -0.05800 ?0.04830 -0.15197410
## MMT00000046 -0.07730 -0.0297 ?1.12e-01 -0.05890 ?0.04430 -0.09380000
## MMT00000051 -0.02260 ?0.0617 -1.29e-01 ?0.08710 -0.11500 -0.06502607
## MMT00000076 -0.00924 -0.1450 ?2.87e-02 -0.04390 ?0.00425 -0.23610000
## MMT00000080 -0.04870 ?0.0582 -4.83e-02 -0.03710 ?0.02510 ?0.08504274
## MMT00000102 ?0.17600 -0.1890 -6.50e-02 -0.00846 -0.00574 -0.01807182
## ? ? ? ? ? ? ? ?F2_23 ? ?F2_24
## MMT00000044 -0.00129 -0.23600
## MMT00000046 ?0.09340 ?0.02690
## MMT00000051 ?0.00249 -0.10200
## MMT00000076 -0.06900 ?0.01440
## MMT00000080 ?0.04450 ?0.00167
## MMT00000102 -0.12500 -0.06820數(shù)據(jù)篩選
## 篩選中位絕對偏差前75%的基因,至少M(fèi)AD大于0.01
## 篩選后會(huì)降低運(yùn)算量,也會(huì)失去部分信息
## 也可不做篩選,使MAD大于0即可
m.mad <- apply(dataExpr,1,mad)
dataExprVar <- dataExpr[which(m.mad >
? ? ? ? ? ? ? ? max(quantile(m.mad, probs=seq(0, 1, 0.25))[2],0.01)),]
## 轉(zhuǎn)換為樣品在行,基因在列的矩陣
dataExpr <- as.data.frame(t(dataExprVar))
## 檢測缺失值
gsg = goodSamplesGenes(dataExpr, verbose = 3)
## ?Flagging genes and samples with too many missing values...
## ? ..step 1
if (!gsg$allOK){
?# Optionally, print the gene and sample names that were removed:
?if (sum(!gsg$goodGenes)>0)
? ?printFlush(paste("Removing genes:",
? ? ? ? ? ? ? ? ? ? paste(names(dataExpr)[!gsg$goodGenes], collapse = ",")));
?if (sum(!gsg$goodSamples)>0)
? ?printFlush(paste("Removing samples:",
? ? ? ? ? ? ? ? ? ? paste(rownames(dataExpr)[!gsg$goodSamples], collapse = ",")));
?# Remove the offending genes and samples from the data:
?dataExpr = dataExpr[gsg$goodSamples, gsg$goodGenes]
}
nGenes = ncol(dataExpr)
nSamples = nrow(dataExpr)
dim(dataExpr)
## [1] ?134 2697
head(dataExpr)[,1:8]
## ? ? ? MMT00000051 MMT00000080 MMT00000102 MMT00000149 MMT00000159
## F2_2 ?-0.02260000 -0.04870000 ?0.17600000 ?0.07680000 -0.14800000
## F2_3 ? 0.06170000 ?0.05820000 -0.18900000 ?0.18600000 ?0.17700000
## F2_14 -0.12900000 -0.04830000 -0.06500000 ?0.21400000 -0.13200000
## F2_15 ?0.08710000 -0.03710000 -0.00846000 ?0.12000000 ?0.10700000
## F2_19 -0.11500000 ?0.02510000 -0.00574000 ?0.02100000 -0.11900000
## F2_20 -0.06502607 ?0.08504274 -0.01807182 ?0.06222751 -0.05497686
## ? ? ? MMT00000207 MMT00000212 MMT00000241
## F2_2 ? 0.06870000 ?0.06090000 -0.01770000
## F2_3 ? 0.10100000 ?0.05570000 -0.03690000
## F2_14 ?0.10900000 ?0.19100000 -0.15700000
## F2_15 -0.00858000 -0.12100000 ?0.06290000
## F2_19 ?0.10500000 ?0.05410000 -0.17300000
## F2_20 -0.02441415 ?0.06343181 ?0.06627665軟閾值篩選
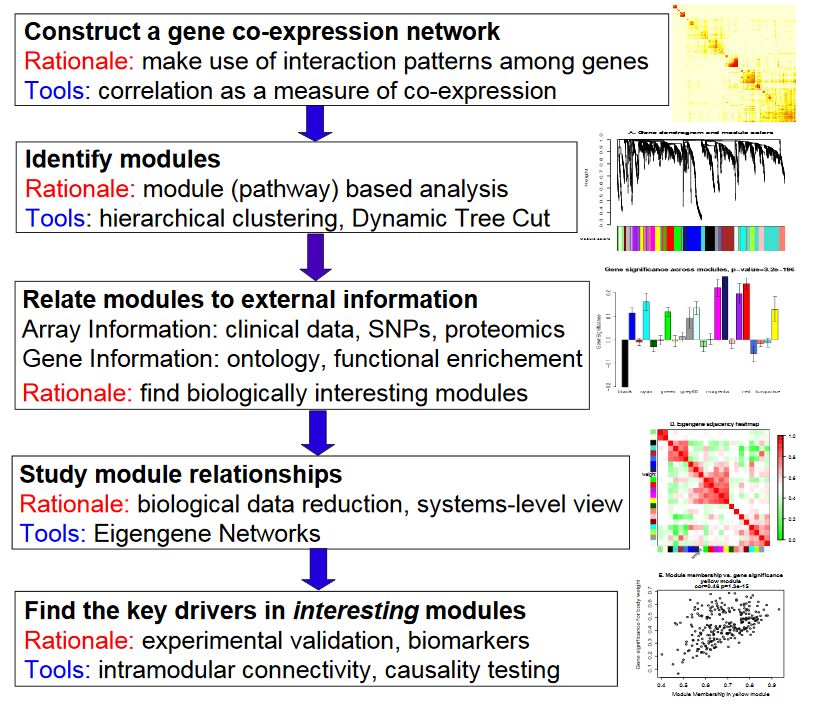
## 查看是否有離群樣品
sampleTree = hclust(dist(dataExpr), method = "average")
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="")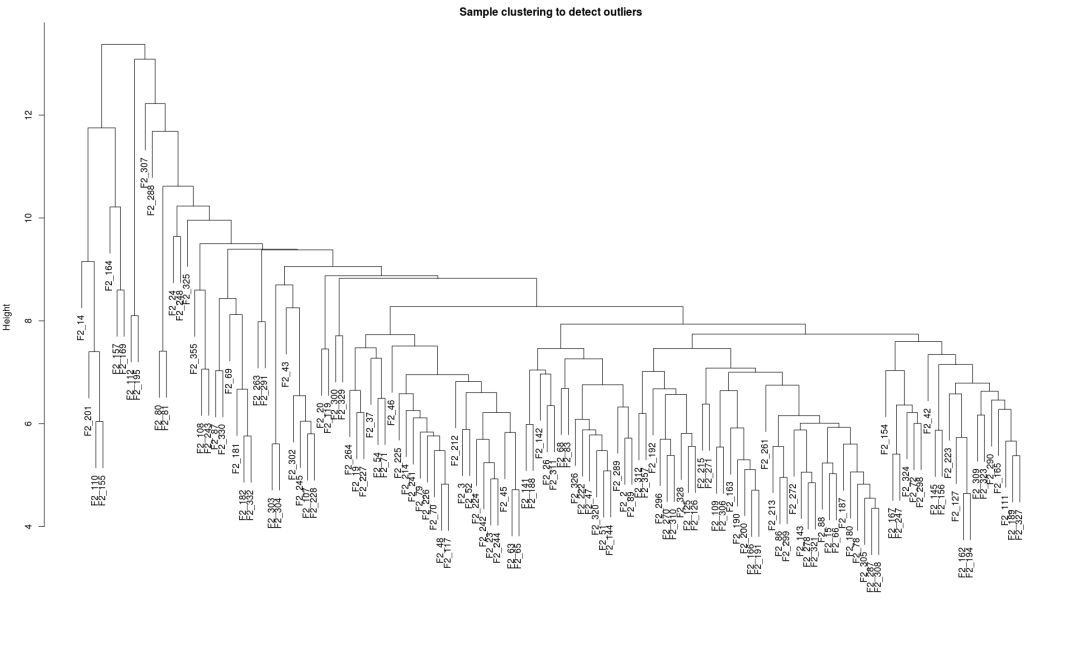
軟閾值的篩選原則是使構(gòu)建的網(wǎng)絡(luò)更符合無標(biāo)度網(wǎng)絡(luò)特征。
powers = c(c(1:10), seq(from = 12, to=30, by=2))
sft = pickSoftThreshold(dataExpr, powerVector=powers,
? ? ? ? ? ? ? ? ? ? ? networkType=type, verbose=5)
## pickSoftThreshold: will use block size 2697.
## pickSoftThreshold: calculating connectivity for given powers...
## ? ..working on genes 1 through 2697 of 2697
## ? Power SFT.R.sq slope truncated.R.sq mean.k. median.k. max.k.
## 1 ? ? 1 ? 0.1370 ?0.825 ? ? ? ? ?0.412 587.000 5.95e+02 922.0
## 2 ? ? 2 ? 0.0416 -0.332 ? ? ? ? ?0.630 206.000 2.02e+02 443.0
## 3 ? ? 3 ? 0.2280 -0.747 ? ? ? ? ?0.920 91.500 8.43e+01 247.0
## 4 ? ? 4 ? 0.3910 -1.120 ? ? ? ? ?0.908 47.400 4.02e+01 154.0
## 5 ? ? 5 ? 0.7320 -1.230 ? ? ? ? ?0.958 27.400 2.14e+01 102.0
## 6 ? ? 6 ? 0.8810 -1.490 ? ? ? ? ?0.916 17.200 1.22e+01 ? 83.7
## 7 ? ? 7 ? 0.8940 -1.640 ? ? ? ? ?0.869 11.600 7.29e+00 ? 75.4
## 8 ? ? 8 ? 0.8620 -1.660 ? ? ? ? ?0.827 ? 8.250 4.56e+00 ? 69.2
## 9 ? ? 9 ? 0.8200 -1.600 ? ? ? ? ?0.810 ? 6.160 2.97e+00 ? 64.2
## 10 ? 10 ? 0.8390 -1.560 ? ? ? ? ?0.855 ? 4.780 2.01e+00 ? 60.1
## 11 ? 12 ? 0.8020 -1.410 ? ? ? ? ?0.866 ? 3.160 9.61e-01 ? 53.2
## 12 ? 14 ? 0.8470 -1.340 ? ? ? ? ?0.909 ? 2.280 4.84e-01 ? 47.7
## 13 ? 16 ? 0.8850 -1.250 ? ? ? ? ?0.932 ? 1.750 2.64e-01 ? 43.1
## 14 ? 18 ? 0.8830 -1.210 ? ? ? ? ?0.922 ? 1.400 1.46e-01 ? 39.1
## 15 ? 20 ? 0.9110 -1.180 ? ? ? ? ?0.926 ? 1.150 8.35e-02 ? 35.6
## 16 ? 22 ? 0.9160 -1.140 ? ? ? ? ?0.927 ? 0.968 5.02e-02 ? 32.6
## 17 ? 24 ? 0.9520 -1.120 ? ? ? ? ?0.961 ? 0.828 2.89e-02 ? 29.9
## 18 ? 26 ? 0.9520 -1.120 ? ? ? ? ?0.944 ? 0.716 1.77e-02 ? 27.5
## 19 ? 28 ? 0.9380 -1.120 ? ? ? ? ?0.922 ? 0.626 1.08e-02 ? 25.4
## 20 ? 30 ? 0.9620 -1.110 ? ? ? ? ?0.951 ? 0.551 6.49e-03 ? 23.5
par(mfrow = c(1,2))
cex1 = 0.9
# 橫軸是Soft threshold (power),縱軸是無標(biāo)度網(wǎng)絡(luò)的評估參數(shù),數(shù)值越高,
# 網(wǎng)絡(luò)越符合無標(biāo)度特征 (non-scale)
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
? ? xlab="Soft Threshold (power)",
? ? ylab="Scale Free Topology Model Fit,signed R^2",type="n",
? ? main = paste("Scale independence"))
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
? ? labels=powers,cex=cex1,col="red")
# 篩選標(biāo)準(zhǔn)。R-square=0.85
abline(h=0.85,col="red")
# Soft threshold與平均連通性
plot(sft$fitIndices[,1], sft$fitIndices[,5],
? ? xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
? ? main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers,
? ? cex=cex1, col="red")
power = sft$powerEstimate
power
## [1] 6經(jīng)驗(yàn)power?(無滿足條件的power時(shí)選用)
# 無向網(wǎng)絡(luò)在power小于15或有向網(wǎng)絡(luò)power小于30內(nèi),沒有一個(gè)power值可以使
# 無標(biāo)度網(wǎng)絡(luò)圖譜結(jié)構(gòu)R^2達(dá)到0.8,平均連接度較高如在100以上,可能是由于
# 部分樣品與其他樣品差別太大。這可能由批次效應(yīng)、樣品異質(zhì)性或?qū)嶒?yàn)條件對
# 表達(dá)影響太大等造成。可以通過繪制樣品聚類查看分組信息和有無異常樣品。
# 如果這確實(shí)是由有意義的生物變化引起的,也可以使用下面的經(jīng)驗(yàn)power值。
if (is.na(power)){
power = ifelse(nSamples<20, ifelse(type == "unsigned", 9, 18),
? ? ? ? ifelse(nSamples<30, ifelse(type == "unsigned", 8, 16),
? ? ? ? ifelse(nSamples<40, ifelse(type == "unsigned", 7, 14),
? ? ? ? ifelse(type == "unsigned", 6, 12)) ? ? ?
? ? ? ? ?)
? ? ? ? ?)
}網(wǎng)絡(luò)構(gòu)建
##一步法網(wǎng)絡(luò)構(gòu)建:One-step network construction and module detection##
# power: 上一步計(jì)算的軟閾值
# maxBlockSize: 計(jì)算機(jī)能處理的最大模塊的基因數(shù)量 (默認(rèn)5000);
# 4G內(nèi)存電腦可處理8000-10000個(gè),16G內(nèi)存電腦可以處理2萬個(gè),32G內(nèi)存電腦可
# ?以處理3萬個(gè)
# ?計(jì)算資源允許的情況下最好放在一個(gè)block里面。
# corType: pearson or bicor
# numericLabels: 返回?cái)?shù)字而不是顏色作為模塊的名字,后面可以再轉(zhuǎn)換為顏色
# saveTOMs:最耗費(fèi)時(shí)間的計(jì)算,存儲(chǔ)起來,供后續(xù)使用
# mergeCutHeight: 合并模塊的閾值,越大模塊越少
net = blockwiseModules(dataExpr, power = power, maxBlockSize = nGenes,
? ? ? ? ? ? ? ? ? ? ? TOMType = type, minModuleSize = 30,
? ? ? ? ? ? ? ? ? ? ? reassignThreshold = 0, mergeCutHeight = 0.25,
? ? ? ? ? ? ? ? ? ? ? numericLabels = TRUE, pamRespectsDendro = FALSE,
? ? ? ? ? ? ? ? ? ? ? saveTOMs=TRUE, corType = corType,
? ? ? ? ? ? ? ? ? ? ? maxPOutliers=maxPOutliers, loadTOMs=TRUE,
? ? ? ? ? ? ? ? ? ? ? saveTOMFileBase = paste0(exprMat, ".tom"),
? ? ? ? ? ? ? ? ? ? ? verbose = 3)
## Calculating module eigengenes block-wise from all genes
## ? Flagging genes and samples with too many missing values...
## ? ? ..step 1
## ..Working on block 1 .
## ? ? TOM calculation: adjacency..
## ? ? ..will use 47 parallel threads.
## ? ? Fraction of slow calculations: 0.000000
## ? ? ..connectivity..
## ? ? ..matrix multiplication (system BLAS)..
## ? ? ..normalization..
## ? ? ..done.
## ? ..saving TOM for block 1 into file WGCNA/LiverFemaleClean.txt.tom-block.1.RData
## ....clustering..
## ....detecting modules..
## ....calculating module eigengenes..
## ....checking kME in modules..
## ? ? ..removing 3 genes from module 1 because their KME is too low.
## ? ? ..removing 5 genes from module 12 because their KME is too low.
## ? ? ..removing 1 genes from module 14 because their KME is too low.
## ..merging modules that are too close..
## ? ? mergeCloseModules: Merging modules whose distance is less than 0.25
## ? ? ? Calculating new MEs...
# 根據(jù)模塊中基因數(shù)目的多少,降序排列,依次編號為 `1-最大模塊數(shù)`。
# **0 (grey)**表示**未**分入任何模塊的基因。
table(net$colors)
##
## ? 0 ? 1 ? 2 ? 3 ? 4 ? 5 ? 6 ? 7 ? 8 ? 9 10 11 12 13
## 135 472 356 333 307 303 177 158 102 94 69 66 63 62層級聚類樹展示各個(gè)模塊
## 灰色的為**未分類**到模塊的基因。
# Convert labels to colors for plotting
moduleLabels = net$colors
moduleColors = labels2colors(moduleLabels)
# Plot the dendrogram and the module colors underneath
# 如果對結(jié)果不滿意,還可以recutBlockwiseTrees,節(jié)省計(jì)算時(shí)間
plotDendroAndColors(net$dendrograms[[1]], moduleColors[net$blockGenes[[1]]],
? ? ? ? ? ? ? ? ? ?"Module colors",
? ? ? ? ? ? ? ? ? dendroLabels = FALSE, hang = 0.03,
? ? ? ? ? ? ? ? ? addGuide = TRUE, guideHang = 0.05)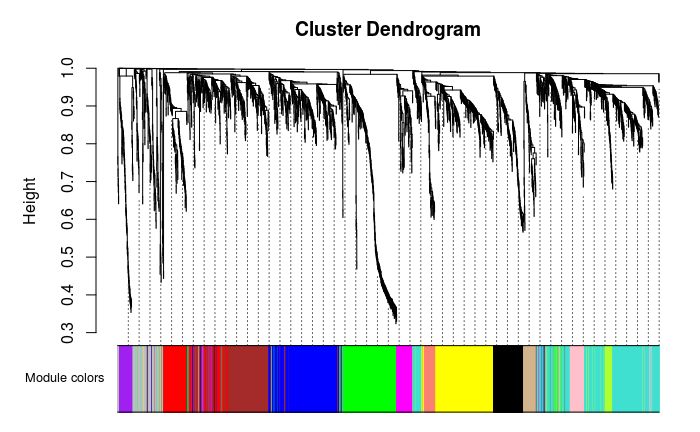
繪制模塊之間相關(guān)性圖
# module eigengene, 可以繪制線圖,作為每個(gè)模塊的基因表達(dá)趨勢的展示
MEs = net$MEs
### 不需要重新計(jì)算,改下列名字就好
### 官方教程是重新計(jì)算的,起始可以不用這么麻煩
MEs_col = MEs
colnames(MEs_col) = paste0("ME", labels2colors(
?as.numeric(str_replace_all(colnames(MEs),"ME",""))))
MEs_col = orderMEs(MEs_col)
# 根據(jù)基因間表達(dá)量進(jìn)行聚類所得到的各模塊間的相關(guān)性圖
# marDendro/marHeatmap 設(shè)置下、左、上、右的邊距
plotEigengeneNetworks(MEs_col, "Eigengene adjacency heatmap",
? ? ? ? ? ? ? ? ? ? ?marDendro = c(3,3,2,4),
? ? ? ? ? ? ? ? ? ? ?marHeatmap = c(3,4,2,2), plotDendrograms = T,
? ? ? ? ? ? ? ? ? ? ?xLabelsAngle = 90)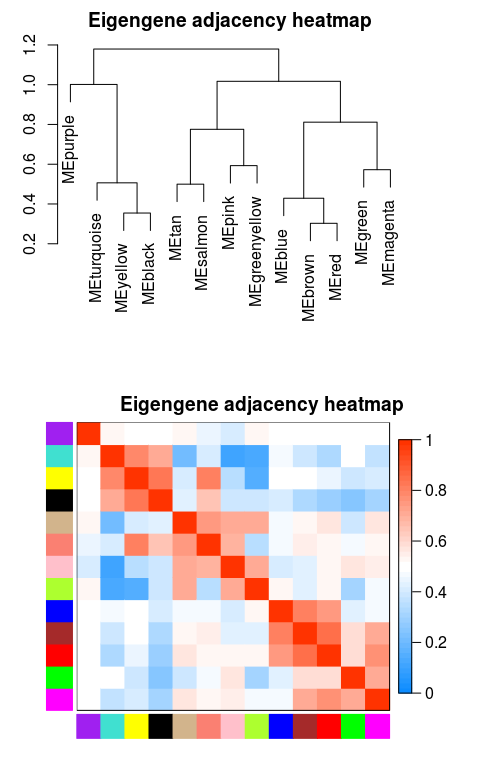
## 如果有表型數(shù)據(jù),也可以跟ME數(shù)據(jù)放一起,一起出圖
#MEs_colpheno = orderMEs(cbind(MEs_col, traitData))
#plotEigengeneNetworks(MEs_colpheno, "Eigengene adjacency heatmap",
# ? ? ? ? ? ? ? ? ? ? ?marDendro = c(3,3,2,4),
# ? ? ? ? ? ? ? ? ? ? ?marHeatmap = c(3,4,2,2), plotDendrograms = T,
# ? ? ? ? ? ? ? ? ? ? ?xLabelsAngle = 90)可視化基因網(wǎng)絡(luò) (TOM plot)
# 如果采用分步計(jì)算,或設(shè)置的blocksize>=總基因數(shù),直接load計(jì)算好的TOM結(jié)果
# 否則需要再計(jì)算一遍,比較耗費(fèi)時(shí)間
# TOM = TOMsimilarityFromExpr(dataExpr, power=power, corType=corType, networkType=type)
load(net$TOMFiles[1], verbose=T)
## Loading objects:
## ? TOM
TOM <- as.matrix(TOM)
dissTOM = 1-TOM
# Transform dissTOM with a power to make moderately strong
# connections more visible in the heatmap
plotTOM = dissTOM^7
# Set diagonal to NA for a nicer plot
diag(plotTOM) = NA
# Call the plot function
# 這一部分特別耗時(shí),行列同時(shí)做層級聚類
TOMplot(plotTOM, net$dendrograms, moduleColors,
? ? ? ?main = "Network heatmap plot, all genes")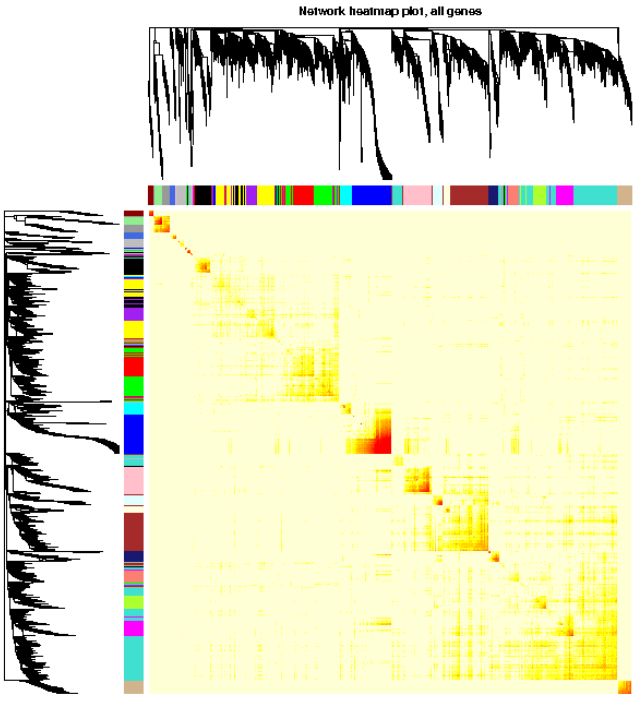
導(dǎo)出網(wǎng)絡(luò)用于Cytoscape
Cytoscape繪制網(wǎng)絡(luò)圖見我們更新版的視頻教程或https://bioinfo.ke.qq.com/。
probes = colnames(dataExpr)
dimnames(TOM) <- list(probes, probes)
# Export the network into edge and node list files Cytoscape can read
# threshold 默認(rèn)為0.5, 可以根據(jù)自己的需要調(diào)整,也可以都導(dǎo)出后在
# cytoscape中再調(diào)整
cyt = exportNetworkToCytoscape(TOM,
? ? ? ? ? ? edgeFile = paste(exprMat, ".edges.txt", sep=""),
? ? ? ? ? ? nodeFile = paste(exprMat, ".nodes.txt", sep=""),
? ? ? ? ? ? weighted = TRUE, threshold = 0,
? ? ? ? ? ? nodeNames = probes, nodeAttr = moduleColors)
關(guān)聯(lián)表型數(shù)據(jù)
trait <- "WGCNA/TraitsClean.txt"
# 讀入表型數(shù)據(jù),不是必須的
if(trait != "") {
?traitData <- read.table(file=trait, sep='\t', header=T, row.names=1,
? ? ? ? ? ? ? ? ? ? ? ? ?check.names=FALSE, comment='',quote="")
?sampleName = rownames(dataExpr)
?traitData = traitData[match(sampleName, rownames(traitData)), ]
}
### 模塊與表型數(shù)據(jù)關(guān)聯(lián)
if (corType=="pearsoon") {
?modTraitCor = cor(MEs_col, traitData, use = "p")
?modTraitP = corPvalueStudent(modTraitCor, nSamples)
} else {
?modTraitCorP = bicorAndPvalue(MEs_col, traitData, robustY=robustY)
?modTraitCor = modTraitCorP$bicor
?modTraitP ? = modTraitCorP$p
}
## Warning in bicor(x, y, use = use, ...): bicor: zero MAD in variable 'y'.
## Pearson correlation was used for individual columns with zero (or missing)
## MAD.
# signif表示保留幾位小數(shù)
textMatrix = paste(signif(modTraitCor, 2), "\n(", signif(modTraitP, 1), ")", sep = "")
dim(textMatrix) = dim(modTraitCor)
labeledHeatmap(Matrix = modTraitCor, xLabels = colnames(traitData),
? ? ? ? ? ? ? yLabels = colnames(MEs_col),
? ? ? ? ? ? ? cex.lab = 0.5,
? ? ? ? ? ? ? ySymbols = colnames(MEs_col), colorLabels = FALSE,
? ? ? ? ? ? ? colors = blueWhiteRed(50),
? ? ? ? ? ? ? textMatrix = textMatrix, setStdMargins = FALSE,
? ? ? ? ? ? ? cex.text = 0.5, zlim = c(-1,1),
? ? ? ? ? ? ? main = paste("Module-trait relationships"))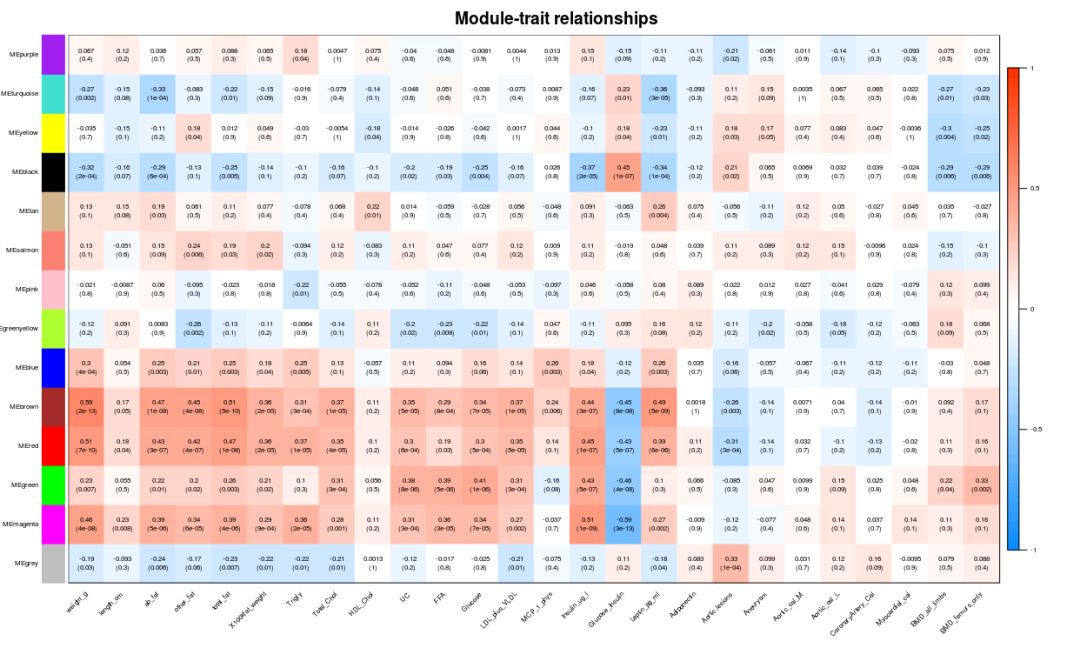
模塊內(nèi)基因與表型數(shù)據(jù)關(guān)聯(lián), 從上圖可以看到MEmagenta與Insulin_ug_l相關(guān),選取這兩部分進(jìn)行分析。
## 從上圖可以看到MEmagenta與Insulin_ug_l相關(guān)
## 模塊內(nèi)基因與表型數(shù)據(jù)關(guān)聯(lián)
# 性狀跟模塊雖然求出了相關(guān)性,可以挑選最相關(guān)的那些模塊來分析,
# 但是模塊本身仍然包含非常多的基因,還需進(jìn)一步的尋找最重要的基因。
# 所有的模塊都可以跟基因算出相關(guān)系數(shù),所有的連續(xù)型性狀也可以跟基因的表達(dá)
# 值算出相關(guān)系數(shù)。
# 如果跟性狀顯著相關(guān)基因也跟某個(gè)模塊顯著相關(guān),那么這些基因可能就非常重要
# 。
### 計(jì)算模塊與基因的相關(guān)性矩陣
if (corType=="pearsoon") {
geneModuleMembership = as.data.frame(cor(dataExpr, MEs_col, use = "p"))
MMPvalue = as.data.frame(corPvalueStudent(
? ? ? ? ? ? as.matrix(geneModuleMembership), nSamples))
} else {
geneModuleMembershipA = bicorAndPvalue(dataExpr, MEs_col, robustY=robustY)
geneModuleMembership = geneModuleMembershipA$bicor
MMPvalue ? = geneModuleMembershipA$p
}
# 計(jì)算性狀與基因的相關(guān)性矩陣
## 只有連續(xù)型性狀才能進(jìn)行計(jì)算,如果是離散變量,在構(gòu)建樣品表時(shí)就轉(zhuǎn)為0-1矩陣。
if (corType=="pearsoon") {
geneTraitCor = as.data.frame(cor(dataExpr, traitData, use = "p"))
geneTraitP = as.data.frame(corPvalueStudent(
? ? ? ? ? ? as.matrix(geneTraitCor), nSamples))
} else {
geneTraitCorA = bicorAndPvalue(dataExpr, traitData, robustY=robustY)
geneTraitCor = as.data.frame(geneTraitCorA$bicor)
geneTraitP ? = as.data.frame(geneTraitCorA$p)
}
## Warning in bicor(x, y, use = use, ...): bicor: zero MAD in variable 'y'.
## Pearson correlation was used for individual columns with zero (or missing)
## MAD.
# 最后把兩個(gè)相關(guān)性矩陣聯(lián)合起來,指定感興趣模塊進(jìn)行分析
module = "magenta"
pheno = "Insulin_ug_l"
modNames = substring(colnames(MEs_col), 3)
# 獲取關(guān)注的列
module_column = match(module, modNames)
pheno_column = match(pheno,colnames(traitData))
# 獲取模塊內(nèi)的基因
moduleGenes = moduleColors == module
sizeGrWindow(7, 7)
par(mfrow = c(1,1))
# 與性狀高度相關(guān)的基因,也是與性狀相關(guān)的模型的關(guān)鍵基因
verboseScatterplot(abs(geneModuleMembership[moduleGenes, module_column]),
? ? ? ? ? ? ? ? ? abs(geneTraitCor[moduleGenes, pheno_column]),
? ? ? ? ? ? ? ? ? xlab = paste("Module Membership in", module, "module"),
? ? ? ? ? ? ? ? ? ylab = paste("Gene significance for", pheno),
? ? ? ? ? ? ? ? ? main = paste("Module membership vs. gene significance\n"),
? ? ? ? ? ? ? ? ? cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)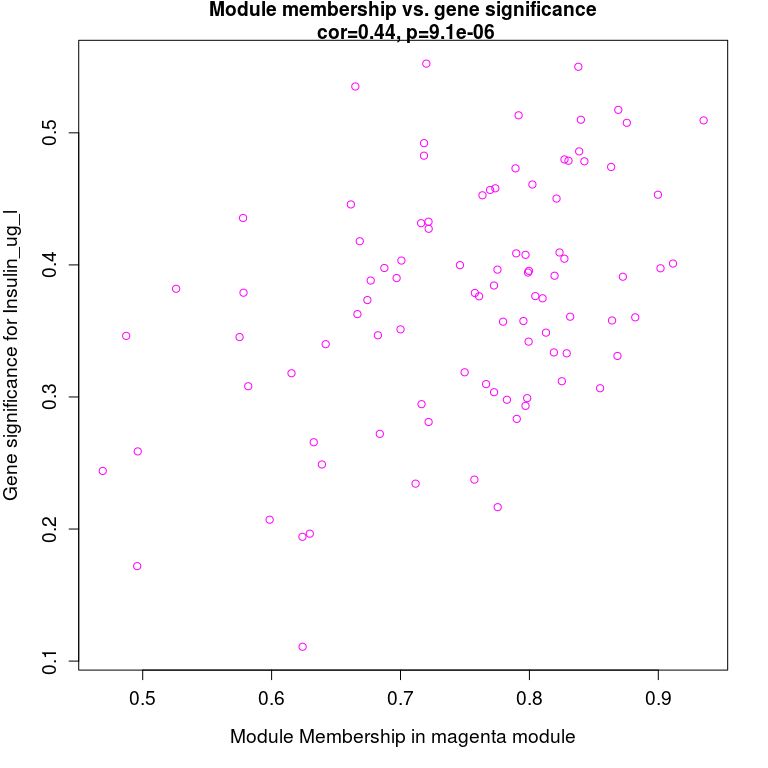
分步法展示每一步都做了什么
### 計(jì)算鄰接矩陣
adjacency = adjacency(dataExpr, power = power)
### 把鄰接矩陣轉(zhuǎn)換為拓?fù)渲丿B矩陣,以降低噪音和假相關(guān),獲得距離矩陣。
TOM = TOMsimilarity(adjacency)
dissTOM = 1-TOM
### 層級聚類計(jì)算基因之間的距離樹
geneTree = hclust(as.dist(dissTOM), method = "average")
### 模塊合并
# We like large modules, so we set the minimum module size relatively high:
minModuleSize = 30
# Module identification using dynamic tree cut:
dynamicMods = cutreeDynamic(dendro = geneTree, distM = dissTOM,
? ? ? ? ? ? ? ? ? ? ? ? ? deepSplit = 2, pamRespectsDendro = FALSE,
? ? ? ? ? ? ? ? ? ? ? ? ? minClusterSize = minModuleSize)
# Convert numeric lables into colors
dynamicColors = labels2colors(dynamicMods)
### 通過計(jì)算模塊的代表性模式和模塊之間的定量相似性評估,合并表達(dá)圖譜相似
的模塊
MEList = moduleEigengenes(datExpr, colors = dynamicColors)
MEs = MEList$eigengenes
# Calculate dissimilarity of module eigengenes
MEDiss = 1-cor(MEs)
# Cluster module eigengenes
METree = hclust(as.dist(MEDiss), method = "average")
MEDissThres = 0.25
# Call an automatic merging function
merge = mergeCloseModules(datExpr, dynamicColors, cutHeight = MEDissThres, verbose = 3)
# The merged module colors
mergedColors = merge$colors;
# Eigengenes of the new merged
## 分步法完結(jié)Reference:
官網(wǎng):https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/
術(shù)語解釋:https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/Simulated-00-Background.pdf
FAQ:https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/faq.html
生信博客:http://blog.genesino.com
你可能還想看
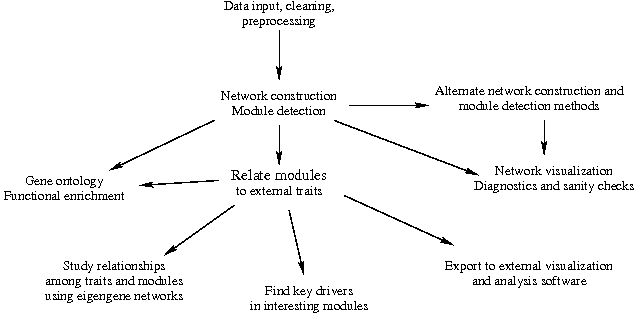
WGCNA教程流程圖:
往期精品(點(diǎn)擊圖片直達(dá)文字對應(yīng)教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后臺回復(fù)“生信寶典福利第一波”或點(diǎn)擊閱讀原文獲取教程合集