前文回顧
1. GATK官方教程 / 概述及工作前的布置
2. GATK教程 / 體細(xì)胞短變異檢測 (SNV+InDel)流程概覽
Data pre-processing for variant discovery
目的
這是第一階段的工作,必須在所有變異發(fā)現(xiàn)之前進(jìn)行。它涉及對原始序列數(shù)據(jù)(以FASTQ或uBAM格式提供)進(jìn)行預(yù)處理,以產(chǎn)生可供分析的BAM文件。 這涉及到對參考基因組的比對,以及一些數(shù)據(jù)清理操作,以糾正技術(shù)偏差,使數(shù)據(jù)適合分析。參考實(shí)現(xiàn)
| | | | |
| | | | |
| uBAM到GVCF或隊(duì)列/cohort VCF | | | |
| | | | |
*
Prod:指的是Broad研究所的數(shù)據(jù)科學(xué)平臺生產(chǎn)管道 (Broad Institute's Data Sciences Platform production pipelines),用于處理Broad的基因組測序平臺設(shè)備產(chǎn)生的序列數(shù)據(jù)。預(yù)期的輸入文件
這個工作流旨在對單個樣本進(jìn)行操作,對于這些樣本,數(shù)據(jù)最初被組織在稱為讀組 (Read groups)的不同子集中。這些相當(dāng)于文庫(即從生物樣本中提取、并準(zhǔn)備進(jìn)行測序的DNA產(chǎn)物,包括使用識別條形碼進(jìn)行片段化和標(biāo)記)與測序通道(即Lane:DNA測序芯片上的物理分隔單元)的“交叉”。此“交叉”是由于多路復(fù)用(即Multiplexing:混合多個庫、并在多個通道上進(jìn)行測序的過程,以降低風(fēng)險(xiǎn)和人為操作所帶來的影響)所產(chǎn)生的。 我們的參考實(shí)踐流程,期望測序數(shù)據(jù)以未比對的BAM (Unmapped BAM, uBAM)文件格式輸入。一些轉(zhuǎn)換程序可將FASTQ轉(zhuǎn)換為uBAM文件。 ① 首先將讀取的序列映射到參考基因組,生成一個按坐標(biāo)排序的SAM/BAM格式的文件。即全基因組比對。 ② 接下來,對重復(fù)序列進(jìn)行標(biāo)記,以減輕一些數(shù)據(jù)生成步驟(如PCR擴(kuò)增)中帶來的偏差。
③ 最后,重新校準(zhǔn)基本質(zhì)量分?jǐn)?shù),因?yàn)?strong>變異調(diào)用算法(Variant calling algorithms)在很大程度上依賴于每個讀序列(Sequence Read)取中分配給單個堿基調(diào)用的質(zhì)量分?jǐn)?shù)(Quality scores)。
映射到參考基因組
涉及的工具:BWA, MergeBamAlignments
http://bio-bwa.sourceforge.net/https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#MergeBamAlignment 第一個處理步驟是逐讀組(Per-read group)執(zhí)行的,包括將每個讀對(Read pair)映射到參考基因組。
參考基因組由公共的基因組序列所合并成的單鏈表示(如人類hg38基因組),旨在為所有基因組分析提供一個共同的坐標(biāo)框架。因?yàn)橛成?比對算法(Mapping algorithm)是單獨(dú)處理每個讀對(Read pair)的,所以可被大規(guī)模并行化,以根據(jù)需要來增加數(shù)據(jù)處理的吞吐量。標(biāo)記重復(fù) (Mark Duplicates) 涉及工具:MarkDuplicatesSpark / MarkDuplicates + SortSam
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#MarkDuplicatesSpark
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#MarkDuplicates
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#SortSam
MarkDuplicatesSpark
此第二個處理步驟是對每個樣本執(zhí)行的,包括識別可能來自相同原始DNA片段的副本(Duplicates/重復(fù))的讀對,這些“Duplicates/重復(fù)”一般是通過一些人工過程(Artifactual processes,如PCR擴(kuò)增和建庫等)。這些被認(rèn)為是非獨(dú)立的觀察,因此程序在每組重復(fù)中標(biāo)記了除單個讀對外的所有讀對,導(dǎo)致標(biāo)記的對(marked pairs)在變異發(fā)現(xiàn)過程中默認(rèn)被忽略。
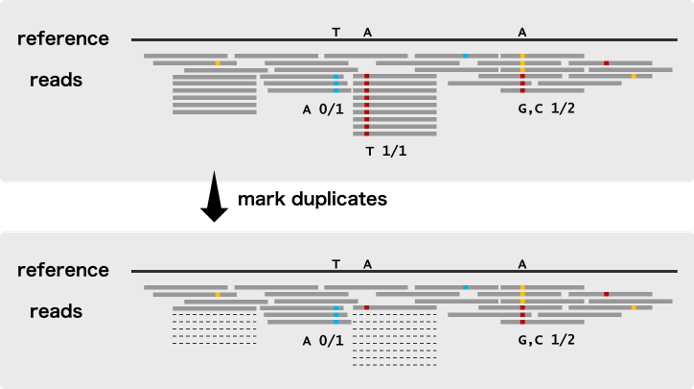
注意上圖中的“虛線”Reads,即為“Duplicates”在測序分析時(shí),duplicate的reads是來源于同一條原始的read,相當(dāng)于是同一個信息,假如某個位置有100條reads覆蓋,90條是duplicate,其實(shí)這個位置就相當(dāng)于90條reads的信息是一個有用信息,如果這個原始read因?yàn)闇y序的問題發(fā)生了一個突變,不考慮duplicate的話,就是90個突變,很容易被作為假陽性檢出 (90/100),而如果考慮是duplicate,這90個read僅被作為一個信息 (1/10),就不太會被檢出了 在這一階段,讀取數(shù)據(jù)還需要按照坐標(biāo)順序進(jìn)行排序,以便進(jìn)行下一步的預(yù)處理。
MarkDuplicatesSpark執(zhí)行重復(fù)標(biāo)記步驟和排序步驟,用于此階段的預(yù)處理。由于在一個樣本中讀取對之間進(jìn)行大量的比對、比較,這一階段的“流水線”(即數(shù)據(jù)處理)一直是一個性能瓶頸,因此MarkDuplicatesSpark利用Apache Spark來并行化進(jìn)程,以便更好地利用所有可用資源。即使不需要訪問專用的Spark集群,這個工具也可以在本地運(yùn)行。
MarkDuplicates + SortSam
作為MarkDuplicatesSpark的替代方案,這個步驟可以通過使用MarkDuplicates的Picard實(shí)現(xiàn),執(zhí)行重復(fù)序列標(biāo)記;然后使用SortSam來對Reads進(jìn)行排序。這兩種工具目前都是作為單線程工具實(shí)現(xiàn)的,因此不能利用核心并行 (Core parallelism)。
建議在擁有大量核心或能夠訪問快速磁盤驅(qū)動器的設(shè)備上,在MarkDuplicatesSpark上運(yùn)行MarkDuplicates,然后運(yùn)行SortSam。如果按照最佳實(shí)踐運(yùn)行,MarkDuplicatesSpark會產(chǎn)生與這種方法相當(dāng)?shù)奈惠敵?(Produces bit-wise equivalent output to this approach),因此上述任何一組工具都是有效的。堿基(質(zhì)量得分)重新校準(zhǔn)/Base (Quality Score) Recalibration 涉及的工具:BaseRecalibrator, Apply
Recalibration, AnalyzeCovariates(可選)
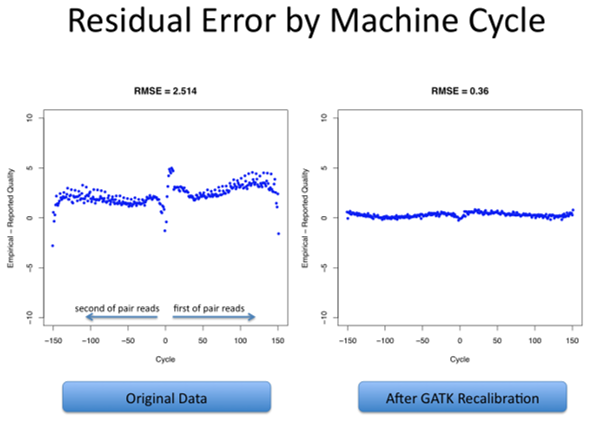
https://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#BaseRecalibratorhttps://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#ApplyBQSRhttps://gatk.broadinstitute.org/hc/en-us/articles/5358824293659--Tool-Documentation-Index#AnalyzeCovariates 這第三個處理步驟是在每個樣本中執(zhí)行的,包括應(yīng)用機(jī)器學(xué)習(xí)來檢測和糾正堿基質(zhì)量分?jǐn)?shù)(Base quality score)中的系統(tǒng)錯誤模式(Patterns of systematic errors),其中堿基質(zhì)量分?jǐn)?shù)是由測序儀對每個堿基給出的置信度分?jǐn)?shù)(Are confidence scores emitted by the sequencer for each base)。 在變異發(fā)現(xiàn)過程中,堿基質(zhì)量分?jǐn)?shù)在權(quán)衡支持或反對可能的變異等位基因(Variant alleles)的證據(jù)方面,發(fā)揮著重要作用,因此糾正數(shù)據(jù)中觀察到的任何系統(tǒng)性偏差是很重要的。 偏差可以來自文庫制備和測序過程中的生化過程,或來自芯片的制造缺陷,又或來自測序儀的儀器缺陷(Instrumentation
defects in the sequencer)。 重新校準(zhǔn)過程包括從數(shù)據(jù)集中的所有堿基調(diào)用(Base calls)中收集協(xié)變量測量值(Covariate measurements),從這些統(tǒng)計(jì)數(shù)據(jù)中構(gòu)建模型,并根據(jù)所得到的模型對數(shù)據(jù)集應(yīng)用堿基質(zhì)量調(diào)整(Base quality adjustments)。 初始統(tǒng)計(jì)數(shù)據(jù)收集可以通過分散在基因組坐標(biāo)上進(jìn)行并行化,通常是通過染色體或染色體批 (Batches)進(jìn)行并行化,但如果需要,可以進(jìn)一步分解以提高數(shù)據(jù)處理的吞吐量。然后每個區(qū)域的統(tǒng)計(jì)數(shù)據(jù)必須被收集到一個單獨(dú)的全基因組范圍內(nèi)的協(xié)變模型(A single genome-wide model of covariation)中;這是不能并行化的,但在計(jì)算上是微不足道的,因此不是性能瓶頸。 最后,將從模型中導(dǎo)出的重新校準(zhǔn)規(guī)則(Recalibration rules)應(yīng)用于原始數(shù)據(jù)集,以生成重新校準(zhǔn)的數(shù)據(jù)集。這與初始統(tǒng)計(jì)數(shù)據(jù)收集在基因組區(qū)域上的并行方式相同,然后進(jìn)行最終文件合并操作,以生成每個樣本的單個分析就緒文件(Analysis-ready file)。一些問題反饋
需要指出的是,Fastq -> Bam過程中,需要包含讀組標(biāo)簽 (Read group tags),因?yàn)樗鼈冊诹鞒痰闹匦滦?zhǔn)堿基質(zhì)量分?jǐn)?shù)階段及其之后需要。
往期精品(點(diǎn)擊圖片直達(dá)文字對應(yīng)教程)
機(jī)器學(xué)習(xí)
后臺回復(fù)“生信寶典福利第一波”或點(diǎn)擊閱讀原文獲取教程合集