讓你的單細(xì)胞數(shù)據(jù)動(dòng)起來(lái)!|iCellR(一)
今天在翻閱single cell 的github時(shí)候,我看見(jiàn)了這個(gè)R包,允許我們處理各種來(lái)自單細(xì)胞測(cè)序技術(shù)的數(shù)據(jù),如scRNA-seq,scVDJ-seq和CITE-Seq。
想看整套的學(xué)習(xí)流程還可以戳這里:
https://vimeo.com/337822487
iCellR優(yōu)點(diǎn)
Single(i)Cell R軟件包(iCellR)在分析pipeline的每個(gè)步驟提供前所未有的靈活性,包括標(biāo)準(zhǔn)化,聚類,降維,插補(bǔ),可視化等。可以通過(guò)無(wú)監(jiān)督和有監(jiān)督聚類進(jìn)行分析, 此外,該工具包提供2D和3D交互式可視化(一個(gè)震撼的交互型3D可視化R包 - 可直接轉(zhuǎn)ggplot2圖為3D),差異表達(dá)分析,基于細(xì)胞、基因和聚類的filter,數(shù)據(jù)合并,dropouts的標(biāo)準(zhǔn)化,數(shù)據(jù)插補(bǔ)方法,校正批次差異,找到標(biāo)記基因的工具聚類和條件,預(yù)測(cè)細(xì)胞類型和偽時(shí)序分析(NBT|45種單細(xì)胞軌跡推斷方法比較,110個(gè)實(shí)際數(shù)據(jù)集和229個(gè)合成數(shù)據(jù)集)。感覺(jué)能做的真的好多啊!
安裝iCellR
# 從鏡像中進(jìn)行安裝
install.packages("iCellR")
# 也可以從github中進(jìn)行安裝
#library(devtools)
#install_github("rezakj/iCellR")
# or
#git clone https://github.com/rezakj/iCellR.git
#R
#install.packages('iCellR/', repos = NULL, type="source")
下載演示數(shù)據(jù)
ion(samples = c("sample1","sample2","sample3"),
# 設(shè)置需要下載的文件夾位置
setwd("/your/download/directory")
# 通過(guò)URL進(jìn)行數(shù)據(jù)下載
sample.file.url = "https://s3-us-west-2.amazonaws.com/10x.files/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz"
# 下載文件
download.file(url = sample.file.url,
destfile = "pbmc3k_filtered_gene_bc_matrices.tar.gz",
method = "auto")
# 解壓縮.
untar("pbmc3k_filtered_gene_bc_matrices.tar.gz")更多的數(shù)據(jù)可以通過(guò)這里獲得:
https://genome.med.nyu.edu/results/external/iCellR/
如何使用iCellR分析scRNA-seq數(shù)據(jù)
1. 加載iCellR包和下載的PBMC樣本數(shù)據(jù)。
library("iCellR")
my.data <- load10x("filtered_gene_bc_matrices/hg19/")
#該目錄有barcodes.tsv,genes.tsv和matrix.mtx文件,將10x的數(shù)據(jù)讀入數(shù)據(jù)框
# 如果是SMART-seq2中有名的小鼠全器官數(shù)據(jù),讀入方式如下
# my.data = read.delim("FACS/Kidney-counts.csv", sep=",", header=TRUE)2. 匯總數(shù)據(jù)
dim(my.data)
# [1] 32738 2700
# 將示例數(shù)據(jù)進(jìn)行拆分為3個(gè)samples
sample1 <- my.data[1:900]
sample2 <- my.data[901:1800]
sample3 <- my.data[1801:2700]
# 合并示例文件my.data <- data.aggregat condition.names = c("WT","KO","Ctrl"))
# 查看數(shù)據(jù)
head(my.data)[1:2]
# WT_AAACATACAACCAC-1 WT_AAACATTGAGCTAC-1
#A1BG 0 0
#A1BG.AS1 0 0
#A1CF 0 0
#A2M 0 0
#A2M.AS1 0 03. 制作iCellR類的對(duì)象。
my.obj <- make.obj(my.data)
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 0.99.0
Raw/original data dimentions (rows,columns): 32738,2700
Data conditions in raw data: Ctrl,KO,WT (900,900,900)
Row names: A1BG,A1BG.AS1,A1CF ...
Columns names: WT_AAACATACAACCAC.1,WT_AAACATTGAGCTAC.1,WT_AAACATTGATCAGC.1 ...
###################################
QC stats performed: FALSE , PCA performed: FALSE , CCA performed: FALSE
Clustering performed: FALSE , Number of clusters: 0
tSNE performed: FALSE , UMAP performed: FALSE , DiffMap performed: FALSE
Main data dimentions (rows,columns): 0 0
Normalization factors: ...
Imputed data dimentions (rows,columns): 0 0
############## scVDJ-Seq ###########
VDJ data dimentions (rows,columns): 0 0
############## CITE-Seq ############
ADT raw data dimentions (rows,columns): 0 0
ADT main data dimentions (rows,columns): 0 0
ADT columns names: ...
ADT row names: ...
######## iCellR object made ########4. 進(jìn)行質(zhì)控(QC)
my.obj <- qc.stats(my.obj) # 計(jì)算每個(gè)細(xì)胞的UMI數(shù)和基因數(shù),還有細(xì)胞周期基因和線粒體基因('mito.percent')的百分比5. 細(xì)胞周期預(yù)測(cè)
和上一個(gè)推送的細(xì)胞周期預(yù)測(cè)方法還是較為一致的,都是通過(guò)評(píng)分進(jìn)行周期的預(yù)測(cè),
my.obj <- cc(my.obj, s.genes = s.phase, g2m.genes = g2m.phase)
head(my.obj@stats)
# CellIds nGenes UMIs mito.percent
#WT_AAACATACAACCAC.1 WT_AAACATACAACCAC.1 781 2421 0.030152829
#WT_AAACATTGAGCTAC.1 WT_AAACATTGAGCTAC.1 1352 4903 0.037935958
#WT_AAACATTGATCAGC.1 WT_AAACATTGATCAGC.1 1131 3149 0.008891712
#WT_AAACCGTGCTTCCG.1 WT_AAACCGTGCTTCCG.1 960 2639 0.017430845
#WT_AAACCGTGTATGCG.1 WT_AAACCGTGTATGCG.1 522 981 0.012232416
#WT_AAACGCACTGGTAC.1 WT_AAACGCACTGGTAC.1 782 2164 0.016635860
# S.phase.probability g2m.phase.probability S.Score
#WT_AAACATACAACCAC.1 0.0012391574 0.0004130525 0.030569081
#WT_AAACATTGAGCTAC.1 0.0002039568 0.0004079135 -0.077860621
#WT_AAACATTGATCAGC.1 0.0003175611 0.0019053668 -0.028560560
#WT_AAACCGTGCTTCCG.1 0.0007578628 0.0011367942 0.001917225
#WT_AAACCGTGTATGCG.1 0.0000000000 0.0020387360 -0.020085210
#WT_AAACGCACTGGTAC.1 0.0000000000 0.0000000000 -0.038953135
# G2M.Score Phase
#WT_AAACATACAACCAC.1 -0.0652390011 S
#WT_AAACATTGAGCTAC.1 -0.1277015099 G1
#WT_AAACATTGATCAGC.1 -0.0036505733 G1
#WT_AAACCGTGCTTCCG.1 -0.0499511543 S
#WT_AAACCGTGTATGCG.1 0.0009426363 G2M
#WT_AAACGCACTGGTAC.1 -0.0680240629 G1
# 繪制細(xì)胞周期圖
pie(table(my.obj@stats$Phase))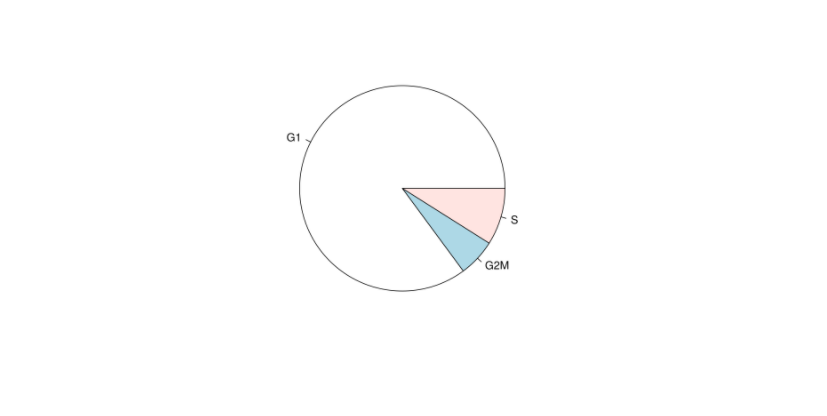
6. Plot QC
默認(rèn)情況下的繪圖功能都將創(chuàng)建交互式html文件,可以設(shè)置interactive = FALSE來(lái)關(guān)閉。
# 繪制 三個(gè)分組中UMIs, genes和 percent mito的beanplot分布圖,想單獨(dú)畫地話可以用`?stats.plot`help一下如何修改參數(shù)
stats.plot(my.obj,
plot.type = "all.in.one",
out.name = "UMI-plot",
interactive = FALSE,
cell.color = "slategray3",
cell.size = 1,
cell.transparency = 0.5,
box.color = "red",
box.line.col = "green")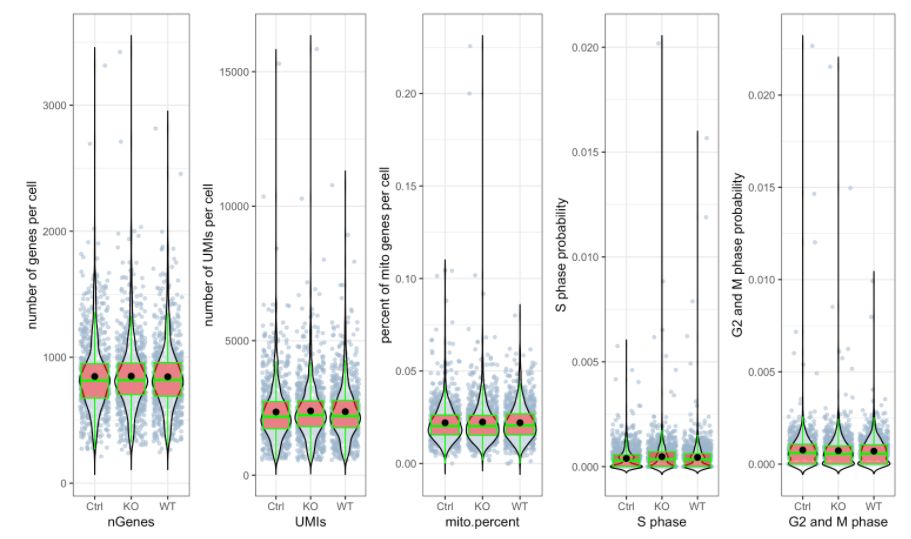
# 繪制散點(diǎn)圖查看線粒體基因和基因分布,注意參數(shù)`plot.type`
stats.plot(my.obj, plot.type = "point.mito.umi", out.name = "mito-umi-plot")###該圖為線粒體基因和UMI的相關(guān)表達(dá)分布(一般原則下會(huì)認(rèn)為線粒體基因比例較高可能為破碎衰老細(xì)胞,可以設(shè)置cutoff值去掉,但如果研究為高代謝或本身耗能較多的細(xì)胞時(shí)需要考慮)
stats.plot(my.obj, plot.type = "point.gene.umi", out.name = "gene-umi-plot")###此圖為UMI和GENE的表達(dá)分布(原則上遵循線性分布)
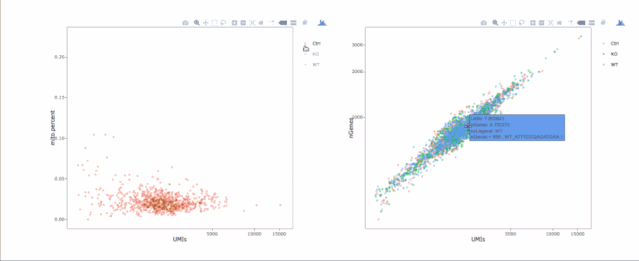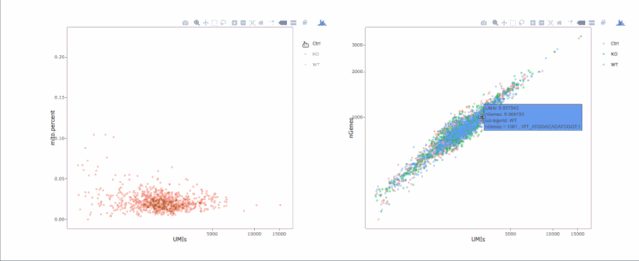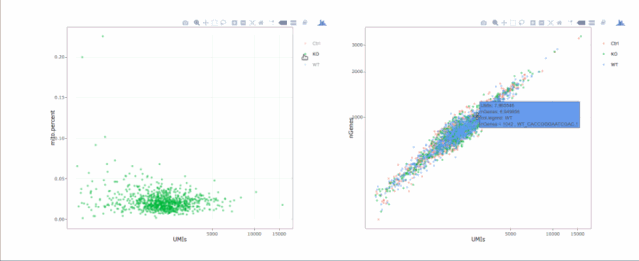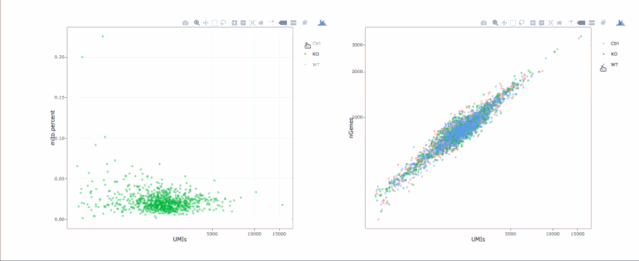
7. 過(guò)濾細(xì)胞
my.obj <- cell.filter(my.obj,
min.mito = 0,
max.mito = 0.05,
min.genes = 200,
max.genes = 2400,
min.umis = 0,
max.umis = Inf)
#[1] "cells with min mito ratio of 0 and max mito ratio of 0.05 were filtered." 保留線粒體基因比例在0到0.05的細(xì)胞
#[1] "cells with min genes of 200 and max genes of 2400 were filtered." 保留基因數(shù)在200到2400之間的細(xì)胞
#[1] "No UMI number filter" 不通過(guò)UMI數(shù)和細(xì)胞ID來(lái)篩選
#[1] "No cell filter by provided gene/genes"
#[1] "No cell id filter"
#[1] "filters_set.txt file has beed generated and includes the filters set for this experiment."篩選之后會(huì)生成一個(gè)文件filter_set,含有實(shí)驗(yàn)的篩選設(shè)置
# more examples 也可以通過(guò)基因和細(xì)胞ID來(lái)篩選
# my.obj <- cell.filter(my.obj, filter.by.gene = c("RPL13","RPL10")) # filter our cell having no counts for these genes
# my.obj <- cell.filter(my.obj, filter.by.cell.id = c("WT_AAACATACAACCAC.1")) # filter our cell cell by their cell ids.
# 查看剩下的細(xì)胞數(shù)(線粒體基因比例在0到0.05,基因數(shù)在200到2400之間)
dim([email protected])
#[1] 32738 2637
# 過(guò)濾掉了63個(gè)細(xì)胞
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(九)- Scater包單細(xì)胞過(guò)濾
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(十)- Scater基因評(píng)估和過(guò)濾
8. 進(jìn)行標(biāo)準(zhǔn)化
可以根據(jù)自己的研究對(duì)數(shù)據(jù)進(jìn)行標(biāo)準(zhǔn)化。還可以使用iCellR以外的工具對(duì)數(shù)據(jù)進(jìn)行標(biāo)準(zhǔn)化,并將數(shù)據(jù)導(dǎo)入iCellR。對(duì)于大多數(shù)單細(xì)胞研究,建議使用“ranked.glsf”標(biāo)準(zhǔn)化。這種標(biāo)準(zhǔn)化化比較適合具有大量零的矩陣。且在合并數(shù)據(jù)(操作參考步驟2:匯總數(shù)據(jù))的情況下,也能對(duì)批次進(jìn)行校正。
my.obj <- norm.data(my.obj,
norm.method = "ranked.glsf",
top.rank = 500) # best for scRNA-Seq
# **更多的標(biāo)準(zhǔn)化方式**
#my.obj <- norm.data(my.obj, norm.method = "ranked.deseq", top.rank = 500)
#my.obj <- norm.data(my.obj, norm.method = "deseq") # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "global.glsf") # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "rpm", rpm.factor = 100000) # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "spike.in", spike.in.factors = NULL)
#my.obj <- norm.data(my.obj, norm.method = "no.norm") # if the data is already normalized
9. 進(jìn)行二次QC
my.obj <- qc.stats(my.obj,which.data = "main.data")
### 該次QC的主要目的就是計(jì)算UMI數(shù)量,每個(gè)細(xì)胞的基因以及每個(gè)細(xì)胞和細(xì)胞周期基因的線粒體基因的百分比。從圖中可以看出其細(xì)胞周期相關(guān)基因表達(dá)較低,線粒體基因表達(dá)比例也較低
stats.plot(my.obj,
plot.type = "all.in.one",
out.name = "UMI-plot",
interactive = F,
cell.color = "slategray3",
cell.size = 1,
cell.transparency = 0.5,
box.color = "red",
box.line.col = "green",
back.col = "white")
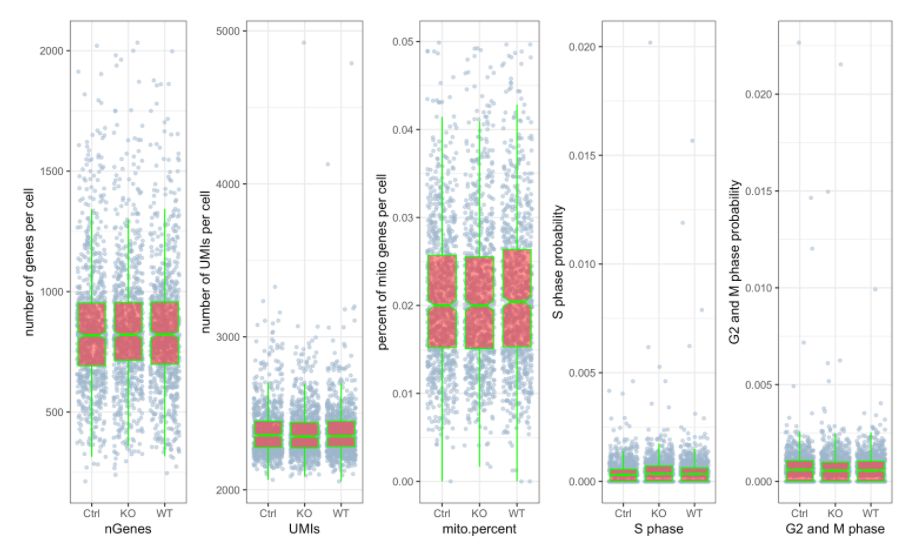
R語(yǔ)言 - 箱線圖(小提琴圖、抖動(dòng)圖、區(qū)域散點(diǎn)圖)
10. Scale data
my.obj <- data.scale(my.obj) # 對(duì)數(shù)據(jù)進(jìn)行中心化和標(biāo)準(zhǔn)化處理11.進(jìn)行g(shù)ene統(tǒng)計(jì)
my.obj <- gene.stats(my.obj, which.data = "main.data")
head([email protected][order([email protected]$numberOfCells, decreasing = T),]) # 將數(shù)據(jù)通過(guò)擁有該基因的細(xì)胞數(shù)按從大到小進(jìn)行排序
# genes numberOfCells totalNumberOfCells percentOfCells meanExp
#30303 TMSB4X 2637 2637 100.00000 38.55948
#3633 B2M 2636 2637 99.96208 45.07327
#14403 MALAT1 2636 2637 99.96208 70.95452
#27191 RPL13A 2635 2637 99.92416 32.29009
#27185 RPL10 2632 2637 99.81039 35.43002
#27190 RPL13 2630 2637 99.73455 32.32106
# SDs condition
#30303 7.545968e-15 all
#3633 2.893940e+01 all
#14403 7.996407e+01 all
#27191 2.783799e+01 all
#27185 2.599067e+01 all
#27190 2.661361e+01 all12.制作聚類的基因模型
在bulk RNA-seq數(shù)據(jù)中,用平均表達(dá)值meanExp排名前500的基因(設(shè)置參數(shù)base.mean.rank對(duì)樣本進(jìn)行聚類是非常常見(jiàn)的,主要原因是為了降低噪聲。在scRNA-seq數(shù)據(jù)中也可以這樣做。并加上ranked.glsf歸一化,會(huì)較為擅長(zhǎng)處理具有大量零的矩陣。
# 看一下高變基因集
make.gene.model(my.obj, my.out.put = "plot",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
no.cell.cycle = T,
out.name = "gene.model")
# 將高變基因集注入`my.obj` 中
my.obj <- make.gene.model(my.obj, my.out.put = "data",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
no.cell.cycle = T,
out.name = "gene.model")
head([email protected]) # 高變基因
# "ACTB" "ACTG1" "ACTR3" "AES" "AIF1" "ALDOA"
# get html plot (optional)
#make.gene.model(my.obj, my.out.put = "plot",
# dispersion.limit = 1.5,
# base.mean.rank = 500,
# no.mito.model = T,
# mark.mito = T,
# interactive = T,
# out.name = "plot4_gene.model")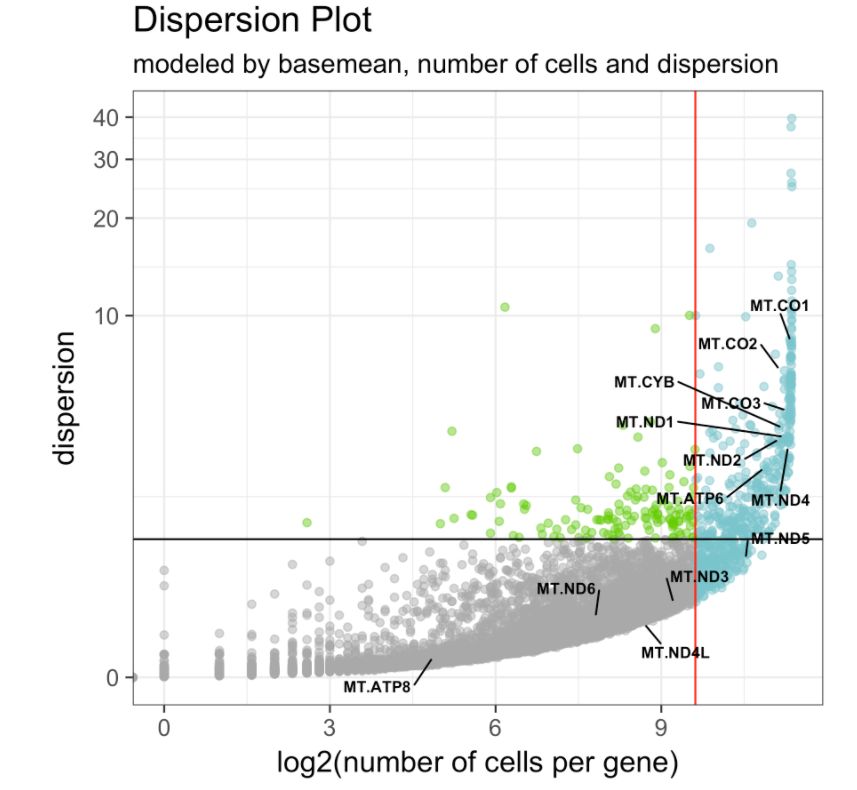
13. 進(jìn)行降維
降維是為了在信息損失較少的情況下,以比原始特征數(shù)量少得多的特征(基因)來(lái)表示數(shù)據(jù),更好地捕捉細(xì)胞之間的關(guān)系,常見(jiàn)降維的方法有PCA,t-SNE,umap和diffusion map。(Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(十一)- Scater單細(xì)胞表達(dá)譜PCA可視化)
PCA分析
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = [email protected],data.type = "main",batch.norm = F)
opt.pcs.plot(my.obj)
##下圖為典型的滾石圖,當(dāng)趨勢(shì)越趨于平緩表明其PC值作用越小,下圖PCA取到10。這一步是可選的,可能在一些情況下也是不適用的。# 2 round PCA (to find top genes in the first 10 PCs and re-run PCA for better clustering
## This is optional and might not be good in some cases
length([email protected])
# 585
my.obj <- find.dim.genes(my.obj, dims = 1:10,top.pos = 20, top.neg = 20) # (optional)
length([email protected])
# 208
# second round PC
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = [email protected],data.type = "main",batch.norm = F)
[email protected]

Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(十一)- Scater單細(xì)胞表達(dá)譜PCA可視化
更多降維方法
# tSNE
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
# UMAP
my.obj <- run.umap(my.obj, dims = 1:10, method = "naive")
# or
# my.obj <- run.umap(my.obj, dims = 1:10, method = "umap-learn")
# diffusion map
# this requires python packge phate
# pip install --user phate
# Install phateR version 2.9
# wget https://cran.r-project.org/src/contrib/Archive/phateR/phateR_0.2.9.tar.gz
# install.packages('phateR/', repos = NULL, type="source")
library(phateR)
my.obj <- run.diffusion.map(my.obj, dims = 1:10, method = "phate")
14. 對(duì)細(xì)胞進(jìn)行聚類分析
“聚類是針對(duì)給定的樣本,依據(jù)它們特征的相似度或距離,將其歸并到若干個(gè)“類”或“簇”的數(shù)據(jù)分析問(wèn)題。一個(gè)類是樣本的一個(gè)子集。直觀上,相似的樣本聚集在相同的類,不相似的樣本分散在不同的類。這里,樣本之間的相似度或距離起著重要作用。”
降維是對(duì)feature (gene) 進(jìn)行操作,而聚類是對(duì)sample進(jìn)行操作,建議在得到高變基因以及降維后操作。(基因表達(dá)聚類分析之初探SOM)
建議使用以下默認(rèn)選項(xiàng):
my.obj <- run.clustering(my.obj,
clust.method = "kmeans", # 聚類方式為k-means
dist.method = "euclidean", # 距離計(jì)算是歐氏距離,有非常明確的空間距離概念
index.method = "silhouette",
max.clust = 25,
min.clust = 2,
dims = 1:10)
# 如果要手動(dòng)設(shè)置cluster數(shù)量,而不使用預(yù)測(cè)的最佳數(shù)量,請(qǐng)將最小值和最大值設(shè)置為所需的數(shù)量:
#my.obj <- run.clustering(my.obj,
# clust.method = "ward.D",
# dist.method = "euclidean",
# index.method = "ccc",
# max.clust = 8,
# min.clust = 8,
# dims = 1:10)
# more examples
#my.obj <- run.clustering(my.obj,
# clust.method = "ward.D",
# dist.method = "euclidean",
# index.method = "kl",
# max.clust = 25,
# min.clust = 2,
# dims = 1:10)
15. 數(shù)據(jù)可視化
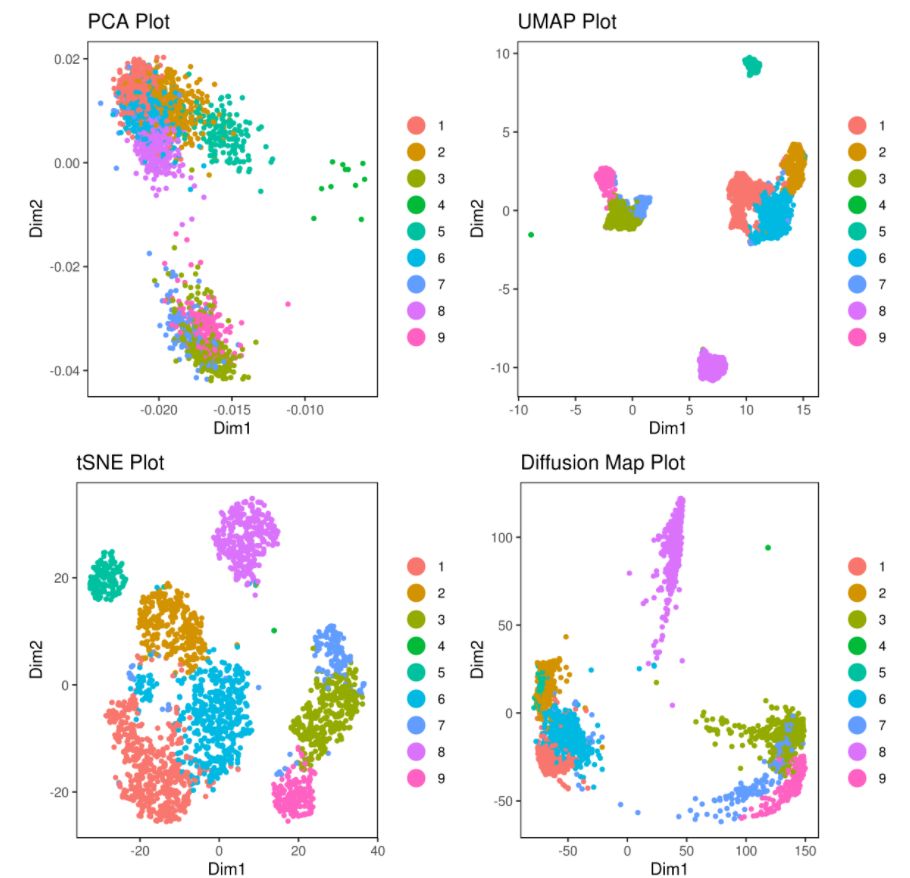
# clusters
A= cluster.plot(my.obj,plot.type = "pca",interactive = F)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F)
D= cluster.plot(my.obj,plot.type = "diffusion",interactive = F)
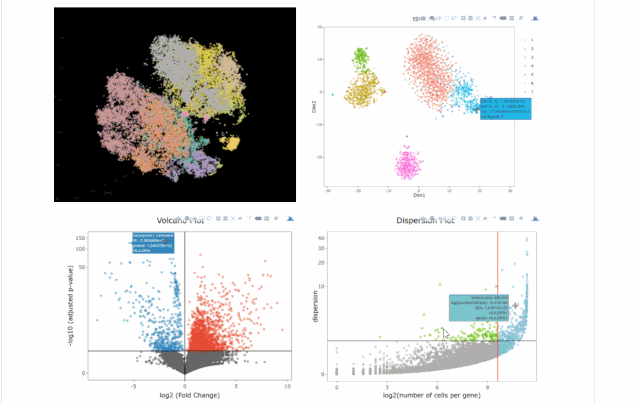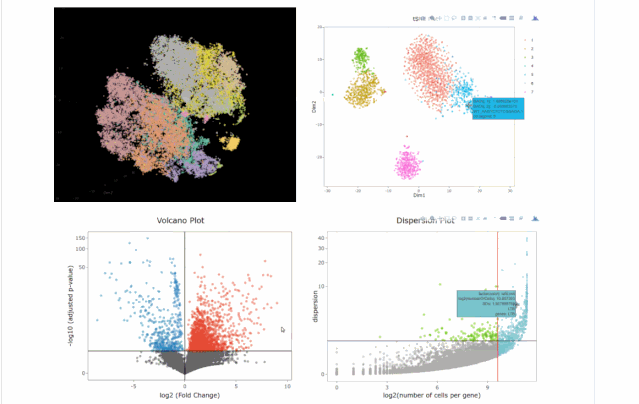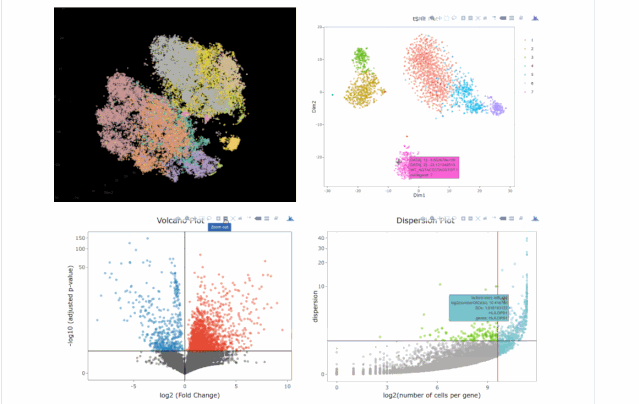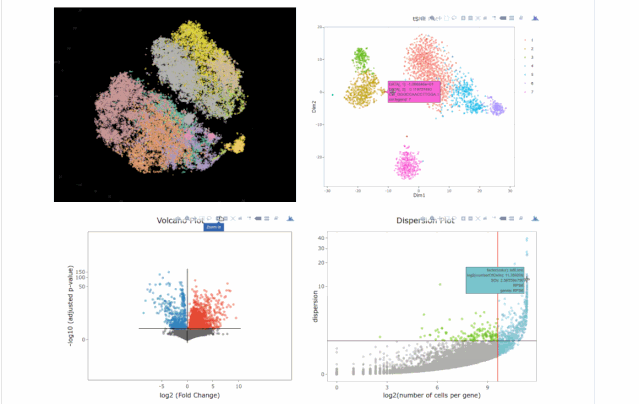
#下圖通過(guò)不同降維方式進(jìn)行展示,其實(shí)我們可以看出,在該組數(shù)據(jù)中還是t-sne降維后的可視化表達(dá)方式最好(細(xì)胞之間的分離度較好)
library(gridExtra)
grid.arrange(A,B,C,D)
# conditions
A= cluster.plot(my.obj,plot.type = "pca",col.by = "conditions",interactive = F)
B= cluster.plot(my.obj,plot.type = "umap",col.by = "conditions",interactive = F)
C= cluster.plot(my.obj,plot.type = "tsne",col.by = "conditions",interactive = F)
D= cluster.plot(my.obj,plot.type = "diffusion",col.by = "conditions",interactive = F)
#在下圖中我們可以發(fā)現(xiàn)無(wú)論哪種降維方式均沒(méi)有batch effect不同條件下的細(xì)胞重合度較好;library(gridExtra)
grid.arrange(A,B,C,D)

16. 三維圖,密度圖和交互式圖
# 2D展示
cluster.plot(my.obj,
cell.size = 1,
plot.type = "tsne",
cell.color = "black",
back.col = "white",
col.by = "clusters",
cell.transparency = 0.5,
clust.dim = 2,
interactive = F)
# 可交互的2D圖
cluster.plot(my.obj,
plot.type = "tsne",
col.by = "clusters",
clust.dim = 2,
interactive = T,
out.name = "tSNE_2D_clusters")
# 可交互的3D圖
cluster.plot(my.obj,
plot.type = "tsne",
col.by = "clusters",
clust.dim = 3,
interactive = T,
out.name = "tSNE_3D_clusters")
# cluster的密度圖
cluster.plot(my.obj,
plot.type = "pca",
col.by = "clusters",
interactive = F,
density=T)
# Density plot for conditions
cluster.plot(my.obj,
plot.type = "pca",
col.by = "conditions",
interactive = F,
density=T)
#下圖為不同可視化方式,盡管方法多種多樣,但個(gè)人感覺(jué)還是2D的方式展示是最為合適的。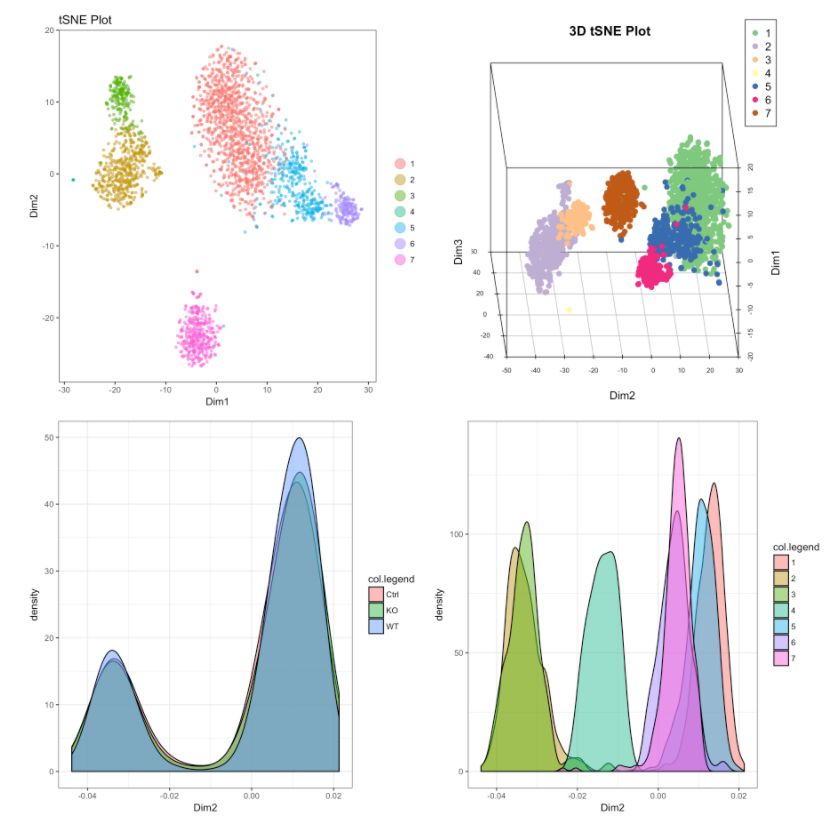
17. 不同cluster和條件下細(xì)胞比例
#如果normalize.ncell = TRUE,它將在三種狀態(tài)(conditions)隨機(jī)**降采樣(down sample)**,因此所有條件都具有相同數(shù)量的細(xì)胞,如果為FALSE則輸出原始細(xì)胞數(shù)。#下圖分別是在不同條件下細(xì)胞的比例關(guān)系,KO,WT,ctrl都是我們較為常見(jiàn)的細(xì)胞分組。# bar plot
clust.cond.info(my.obj, plot.type = "bar", normalize.ncell = FALSE, my.out.put = "plot")
# Pie chart
clust.cond.info(my.obj, plot.type = "pie", normalize.ncell = FALSE, ,my.out.put = "plot")
# data
my.obj <- clust.cond.info(my.obj, plot.type = "bar", normalize.ncell = F)
#head([email protected])
# conditions clusters Freq
#1 ctrl 1 199
#2 KO 1 170
#3 WT 1 182
#4 ctrl 2 106
#5 KO 2 116
#6 WT 2 113
到這里,我們其實(shí)已經(jīng)表現(xiàn)了許多的plot,下期接著介紹吧!
單細(xì)胞
Science:通過(guò)單細(xì)胞轉(zhuǎn)錄組測(cè)序揭示玉米減數(shù)分裂進(jìn)程 | 很好的單細(xì)胞分析案例
Nature系列 整合單細(xì)胞轉(zhuǎn)錄組學(xué)和質(zhì)譜流式確定類風(fēng)濕性關(guān)節(jié)炎滑膜組織中的炎癥細(xì)胞狀態(tài) 詳細(xì)解讀
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(二)- 實(shí)驗(yàn)平臺(tái)
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(三)- 原始數(shù)據(jù)質(zhì)控
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(四)- 文庫(kù)拆分和細(xì)胞鑒定
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(五)- STAR, Kallisto定量
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(六)- 構(gòu)建表達(dá)矩陣,UMI介紹
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(七)- 導(dǎo)入10X和SmartSeq2數(shù)據(jù)Tabula Muris
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(八)- Scater包輸入導(dǎo)入和存儲(chǔ)
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(九)- Scater包單細(xì)胞過(guò)濾
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(十)- Scater基因評(píng)估和過(guò)濾
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(十一)- Scater單細(xì)胞表達(dá)譜PCA可視化
Hemberg-lab單細(xì)胞轉(zhuǎn)錄組數(shù)據(jù)分析(十二)- Scater單細(xì)胞表達(dá)譜tSNE可視化
植物單細(xì)胞轉(zhuǎn)錄組的春天來(lái)了,還不上車?Science, PC, PP, MP, bioRxiv各一個(gè)
高顏值免費(fèi)在線繪圖
往期精品(點(diǎn)擊圖片直達(dá)文字對(duì)應(yīng)教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后臺(tái)回復(fù)“生信寶典福利第一波”或點(diǎn)擊閱讀原文獲取教程合集